
Abstract
Chromohalobacter salexigens DSM 3043 can grow over a wide range of salinity, which makes it as an excellent model organism for understanding the mechanism of prokaryotic osmoregulation. Functional analysis of C. salexigens genes is an essential way to reveal their roles in cellular osmoregulation. However, the lack of an effective markerless gene deletion system has prevented construction of multiple gene deletion mutants for the members in the genus. Here, we report the development of a markerless gene deletion system in C. salexigens using allelic exchange method. In this system, the in vitro mutant allele of target gene was inserted into a pK18mobsacB-based integrative vector pMDC21, which contained a chloramphenicol resistance cassette as the positive selection marker and a sacB gene from Bacillus subtilis as the counterselectable marker. To validate this system, two single-gene deletion mutants and a double-gene deletion mutant were constructed. In addition, our results showed that growth of the merodiploids and sucrose screening at 25 °C were more effective to decrease the occurrence of spontaneous sucrose resistance colonies than at higher temperature (30 or 37 °C), and growth of the merodiploids in mineral salt medium instead of the complex medium was critical to increase the recovery rate of deletion mutants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chromohalobacter salexigens DSM 3043 is a metabolically versatile, moderately halophilic bacterium that can grow over a wide range of salinity, in the presence of NaCl concentrations ranging from 0.5 to 3 M NaCl in M63 glucose-defined medium and from 0.1 to 4 M NaCl in complex medium, which makes it as excellent model organism for studying prokaryotic osmoadaptation (Arahal et al. 2001; Canovas et al. 1996; Vargas et al. 2008; Vreeland et al. 1980) In addition, C. salexigens strains have recently received a considerable interest as resources for ectoine, hydroxyectoine, and poly-hydroxybutyrate production (Vargas and Nieto 2004; Ventosa and Nieto 1995; Yin et al. 2015). Because most of them are very easy to grow and maintain in the laboratory and their nutritional requirements are very simple, increasing effects have been made to use halophiles as cell factories for bioprocessing with advantages of the lower energy and less fresh water consumption (Yin et al. 2015).
The complete genome sequence of C. salexigens DSM 3043 had been determined by the Joint Genome Institute of the US Department of Energy (Copeland et al. 2011), which presents an opportunity for understanding its characteristic metabolic network that enable it to grow in the presence of high concentrations of NaCl inhibitory to most non-halophiles. To determine gene function, efficient methods for generating mutants are needed. Besides of physical radiation and/or chemical reagents, two biological methods have been employed for the construction of C. salexigens mutants. The first is random mutagenesis with transposon Tn1732 (Canovas et al. 1997; Kunte and Galinski 1995). Tn1732 is a derivative of Tn1721, consisting of the “basic transposon” of Tn1721 that encodes the transposition functions (transposase, tnpA, and resolvase, tnpR, genes). The suicide vector pSUP102-Gm carrying Tn1732 could be delivered into C. salexigens via E. coli SM10 mediated conjugation to disrupt the target genes in the chromosome of C. salexigens. In our laboratory, the suicide plasmid pUTminiTn5 (Manilla-Pérez et al. 2010) carrying miniTn5 was used for insertional mutagenesis of C. salexigens via E. coli SM10 mediated conjugation. Although several salt-sensitive mutants were obtained, the sequence analysis results showed that the distribution of interrupted genes was limited, indicating the presence of “hot spots” for Tn5 integration in C. salexigens (unpublished results). Other transposons belonging to the Tn7 and Tn10 families had been used for insertional mutagenesis in C. salexigens, but it was unsuccessful (Argandoña et al. 2012). These results cause severe limitation to the use of transposons in C. salexigens. The second approach is targeted mutagenesis by allelic exchange between the target genes and in vitro-modified alleles which can be manipulated by introducing deletions, insertions or base replacements. In C. salexigens, the knockout mutants reported were usually generated by antibiotic marker allelic exchange (García-Estepa et al. 2006; Reina-Bueno et al. 2012; Rodríguez-Moya et al. 2010; Salvador et al. 2015; Tokunaga et al. 2004). No matter what kinds of strategies used as mentioned above, a cassette containing antibiotic resistance gene was ultimately retained in the modified organism, which may result in some limitations in complementation analysis or in the construction of multiple deletion mutations in a strain. Because the number of available antibiotic markers that could be used for halophiles is much lower than that for non-halophiles, multiple gene deletion in C. salexigens with marker exchange method is extremely difficult. Besides that, insertion of antibiotic resistance cassettes may cause polar effects on the expression of downstream genes at the target locus, especially for the polycistronic operons. In order to overcome this drawback, a more efficient method to delete target genes or genomic regions without leaving selection markers or foreign DNA sequences behind should be established.
Recently, CRISPR (clustered regularly interspaced short palindromic repeats)-Cas9 system, a novel gene editing tool, has been successfully applied to gene targeting in microorganisms, plants and animals (Bortesi et al. 2016). This system usually has two main components, Cas9-nuclease and a single chimeric guide RNA (sgRNA), and can modify multiple genes in the chromosome at one time with the short protospacer sequences of the sgRNA (Ran et al. 2013). To apply this technique for the genome editing of C. salexigens DSM 3043, the main technical barrier to be resolved is how to transform the sgRNA into C. salexigens cells. However, the methods for direct transformation of DNA or RNA fragments into C. salexigens cells by electroporation or other treatments are not available now, which make it impossible for construction of C. salexigens mutants with this system at the current situation. In this report, we described using the suicide vector pMDC21, a derivative of pK18mobsacB to generate unmarked, in-frame deletion mutants in C. salexigens via allelic exchange. To demonstrate the reliability of our system, two single-gene deletion mutants and one double-gene deletion mutant of C. salexigens DSM 3043 were constructed, and the factors to reduce the ratio of the spontaneous sucrose resistance colonies and improve the recovery rate of desired recombinants were investigated as well.
Materials and methods
Strains, plasmids, primers, and growth conditions
Bacterial strains and plasmids used in this work are listed in Table 1. Escherichia coli DH5α and S17-1 strains were routinely grown at 37 °C under aerobic condition in Luria–Bertani (LB) medium (per liter: tryptone 10 g, yeast extract 5 g, NaCl 10 g, pH 7.5) (Sambrook et al. 1989), or in M63 medium (per liter: (NH4)2SO4 2 g, KH2PO4, 13.6 g, FeSO4·7H2O 0.5 mg, MgSO4, 0.12 g, d-glucose 3.6 g, adjust pH to 7.5 with 10 M KOH). C. salexigens DSM 3043 and its derivative strains were routinely grown at 37 °C with aeration in D5 medium (per liter: NaCl 50 g, MgSO4·7H2O 5 g, tryptone 5 g, and yeast extract 3 g, pH 7.5), or in M63 medium (pH 7.5) supplemented with 0.86 M NaCl. Solid media were obtained by adding 20 g of agar powder (DaMao, TianJin, China) per liter. LB medium containing 4 mM MgSO4 and additional 0.35 M NaCl was named as LB-Mg medium, while LB medium lacking the ingredient NaCl was named as YET medium. When required, the media were supplemented with the substrates and antibiotics at the following concentrations: 20 mM glycine betaine, 20 mM dimethylglycine (DMG), 20 mM sarcosine, 20 mM glycine, 20 mM glucose, 292 mM sucrose, 70 μg ml−1 chloramphenicol (Cm), 50 μg ml−1 kanamycin (Km), and 50 μg ml−1 rifampicin (Rif).
General DNA manipulations
Routine DNA manipulations were carried out as described previously (Sambrook et al. 1989). Genomic and plasmid DNAs were extracted from bacteria using the Ezup Column Bacteria Genomic DNA Extraction Kit (Sangon, China) and SanPrep Column Plasmid Mini-Preps Kit (Sangon, China), respectively. After electrophoresis, DNA fragments were purified using the SanPrep Column DNA Gel Extraction Kit (Sangon, China). T4 DNA ligase and restriction enzymes were purchased from TaKaRa Biotechnology Co., Ltd. (Dalian, China) and used as recommended by the manufacturer’s instructions. Amino acid and DNA alignments were done using the software DNAMAN version 8.0 (Lynnon Biosoft). Polymerase chain reaction (PCR) was normally conducted in 20 μl volume using the Pfu PCR MasterMix (TianGen, China) according to the manufacturer’s instructions. To perform a colony PCR, a small amount of bacterial colony were resuspended in 20 μl of double-distilled water, boiled for 5 min, and centrifuged at 6000×g for 5 min. The resulting supernatant was used as template in the following PCR reaction.
Plasmids construction
The primers used in this study are listed in Table 2, and all DNA sequences used are retrieved from the NCBI web page (https://www.ncbi.nlm.nih.gov/). All of the plasmid regions obtained by PCR were sequenced to verify that no mutations were introduced.
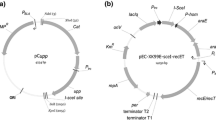
The suicide or integrative plasmid was constructed with pK18mobsacB as the backbone. The Cm resistance cassette was PCR amplified from pACYCDuet-1 using the primers Cm-F and Cm-R. The resulting 1054-bp PCR product was gel-purified, and cloned into BglII-NcoI double-digested pK18mobsacB using the ClonExpress® II One Step Cloning Kit (Vazyme, Nanjing, China), creating pMDC21 (GenBank accession number: KY406738) (Fig. 1).

Plasmid map of the integrative vector pMDC21. pMDC21 has the following features: a pMB1-based origin of replication (rep_ori, suicide plasmid for C. salexigens), the origin of transfer for the RP4 plasmid (oriT_RP4, for conjugal transfer between E. coli and C. salexigens), the pUC18 multiple cloning site (MCS, the mutated allele to be introduced into C. salexigens can be inserted into this site), lacZ alpha (encoding for α-peptide of galactosidase), cat (chloramphenicol resistance gene, positive selection marker), sacB (levansucrase-encoding gene from B. subtilis, negative selection marker). The rep_ori, lacZα, cat and sacB are represented by arrows showing the direction of replication or transcription, while MCS and oriT_RP4 (origin of transfer for the RP4 plasmid) are represented by a bar
The open reading frame (ORF) Csal_1415 encodes a putative Na+/H+ antiporter (NhaC) in C. salexigens DSM 3043 (NCBI reference sequence: WP_011506715.1). pMDCΔcsal_1415 was constructed for the csal_1415 in-frame deletion in strain ZW4-1. Two primer pairs (primers 1415-uF and 1415-uR; primers 1415-dF and 1415-dR) were used to amplify the upstream homologous region (UHR) and downstream homologous region (DHR) of ORF csal_1415 with C. salexigens ZW4-1 genomic DNA as template. The two PCR amplicons, both 955-bp in length, were then fused together by splicing by overlap extension PCR (SOE-PCR) (Horton 1997). For this purpose, the 3′ end of the upstream fragment 1415-uR include a 20-bp overhang with homology to the 5′ end of the primer 1415-dF. The resulting PCR fragment, 1890-bp in length, was gel-purified, double-digested with EcoRI and BamHI, and ligated into the EcoRI and BamHI sites of pMDC21, yielding pMDCΔcsal_1415.
pMDCΔsarC was constructed for the sarC (csal_0998-1001) in-frame deletion in strain ZW4-1. Two pairs of primers (primers sar_uF and sar_uR; primers sar_dF and sar_dR) were used to amplify the UHR (961 bp) and DHR (958 bp) of the sarC gene (encoding for a putative sarcosine oxidase), respectively. The two amplicons were then fused together by SOE-PCR with primers sar_uF and sar_dR to generate a 1899-bp fragment, which was gel-purified, double-digested with EcoRI and BamHI, and ligated to pMDC21, yielding pMDCΔsarC.
Generation of single- and double-gene deletion mutants
C. salexigens ZW4-1 csal_1415 unmarked, in-frame deletion mutant was constructed by a two-step process: allelic integration, and recombinative excision. First, the allelic exchange plasmid pMDCΔcsal_1415 was transformed into competent E. coli S17-1 cell by the routine CaCl2 method (Sambrook et al. 1989), and subsequently transferred into C. salexigens ZW4-1 strain by biparental filter mating, which were performed as follows. The donor E. coli S17-1 strain harboring pMDCΔcsal_1415 was aerobically grown at 37 °C in LB broth supplemented with chloramphenicol until the late exponential growth phase, and the recipient ZW4-1 strain was grown in D5 medium containing rifampicin until the early stationary phase under the same culture conditions. Both E. coli and C. salexigens cells were separately harvested by centrifugation (5400×g, 5 min), washed two times with 10 mM MgSO4, resuspended in the same solution, and mixed at a ratio of 2:1. Then, 50 μl (equivalent to approximately 109 cells) of the mating mixtures was placed onto a nitrocellulose filter disk (0.45 μm pore size, 25 mm diameter) that had been placed aseptically on LB-Mg agar plate. After incubation at 37 °C for 12–16 h, the cells on the filter disk were resuspended in 500 μl of 10 mM MgSO4, serially diluted, and plated on D5 plates plus Rif (to eliminate the donor bacteria) and Cm (for selection of transconjugants with plasmid integration by the single homologous recombination event). After incubation for 48 h at 37 °C in the dark, the colonies growing on the plates were selected. Colony PCR was performed with the primers sacB-F and sacB-R to detect the plasmid backbone. For the second step, one of the merodiploids was inoculated in D5 medium without antibiotics for 18 h at 30 °C. To select for bacteria that had undergone a second recombinant event, the culture was serially diluted, spread onto D5 plates supplemented with sucrose, and incubated at 30 °C for 48 h. The colonies conferring sucrose resistance were transferred to D5 plates containing Cm by toothpicks. The SucrCms (r: resistant; s: sensitive) colonies carrying either the wild type or mutant allele in the chromosome were randomly selected to perform colony PCR for screening the deletion mutants using two primer sets (sacB-F/sacB-R, 1415-uF/1415-dR). The primers (sacB-F/sacB-R) were used to verify excision of the suicide vector, while the primers (1415-uF/1415-dR) were used to screen the mutant allele in C. salexigens. In addition, PCR tests with an external primer 1415-test-F (anchoring upstream of integration site) and an internal primer 1415-test-R (anchoring upstream homologous region of csal_1415 mutant allele) were used for detection of the site-specific plasmid excision event with the wild-type genomic DNA served as control. Positive colonies were further purified and confirmed by PCR and DNA sequencing. One of the positive clones was designated as C. salexigens 3043Δcsal_1415. A schematic diagram for construction of the csal_1415 in-frame deletion mutant is shown in Fig. 2.

Schematic diagram of allelic exchange procedures for the construction of C. salexigens Δcsal_1415 mutant. a Two homologous regions (UHR and DHR, ~1 kb each) of ORF csal_1415 was cloned into the integrative vector pMDC21, yielding pMDCΔcsal_1415. b The allele replacement vector pMDCΔcsal_1415 was transferred into C. salexigens ZW4-1 by conjugation and integrated into the target locus via one of the homologous regions (UHR or DHR) through the first homologous recombination (HR) event, resulting in Cmr merodiploids. The first HR events are represented by solid lines (through the upstream homologous region, UHR) and dashed lines (through the downstream homologous region n, DHR). Two possible genotypes for the merodiploids are follows: a genotype of the merodiploid arising by the first HR through the UHR; b genotype of the merodiploid arising by the first HR through the DHR. c The merodiploid was grown in M63 medium without antibiotic pressure to induce the second crossover which resulted in the loss of the integrated plasmid, then the culture was plated on D5 plates containing sucrose with and without Cm, and incubated at 25 °C. The colonies exhibiting SucrCms phenotype were picked up and screened for the deletion mutants by colony PCR. If the second HR event occurs on the same side as the first HR event, the wild-type genotype is restored (c). If the second HR event occurs on the opposite side, the wild-type allele is replaced by the mutant allele (d)
A similar approach was used to generate the sarC gene in-frame deletion mutant of C. salexigens ZW4-1. pMDCΔsarC was used as the allelic exchange vector. Transconjugants resulting from a single crossover event via homologous recombinant were verified with the primers sacB-F and sacB-R. To identify ΔsarC mutants, three primer sets were employed in colony PCR detection. Primers sacB-F and sacB-R were used to verify loss of the suicide vector, and primers sarC-uF and sarC-dR were used to verify sarC gene deletion in C. salexigens, while primers sar-test-F and sar-test-R were used for detection of the site-specific plasmid excision event. One of the positive clones was designated as C. salexigens 3043ΔsarC.
For the construction of csal_1415 and sarC double-gene in-frame deletion mutant, pMDCΔsarC was used as allele replacement vector, and C. salexigens 3043Δcsal_1415 strain was used as the recipient cell in conjugation experiment. The primers used for verifying the first and second recombination events, as well as confirmation of the gene deletion, were the same as those used in construction of C. salexigens 3043ΔsarC. One of the mutant was designated as C. salexigens 3043Δcsal_1415ΔsarC.
Growth characteristics of wild type versus mutant of C. salexigens
To measure the growth difference between wild type and Δcsal_1415 mutant, C. salexigens DSM 3043, ZW4-1, 3043Δcsal_1415 and 3043Δcsal_1415ΔsarC strains were individually inoculated to D5 liquid medium, and incubated at 37 °C until early stationary phase. Then, the cells were collected by centrifugation, washed with 10 mM MgSO4 for three times, and resuspended in M63 medium. Subsequently, 1% (v/v) cell suspension, calibrated with growth medium to OD600 = 1.0, were inoculated into M63 media supplemented with 5, 10, and 15% (w/v) NaCl, and the culture was incubated at 37 °C with shaking. Growth was monitored by measuring the optical density of the culture at 600 nm (OD600).
To determine the ability of wild type, ΔsarC and Δcsal_1415ΔsarC mutant strains using glycine betaine, DMG, sarcosine and glycine as their sole carbon source, C. salexigens DSM 3043, ZW4-1, 3043ΔsarC, and 3043Δcsal_1415ΔsarC strains were streaked on M63 agar plates containing 0.86 M NaCl with glucose, glycine betaine, DMG, sarcosine or glycine as the sole carbon source, and cultivated at 37 °C for 72 h.
Results and discussion
Choosing the suitable selection markers
The method of allelic exchange between a target gene in the chromosomal DNA and a mutated allele carried by a suicide plasmid, was used to construct the unmarked deletion mutants of C. salexigens. The suicide plasmid normally harbored an antibiotic-resistant marker and a counterselectable marker such as sacB, rpsL or tetAR (Reyrat et al. 1998). The sacB gene from Bacillus subtilis encodes a levansucrase, which is lethal in most Gram-negative bacteria in the presence of sucrose (Gay et al. 1983; Reyrat et al. 1998). Salvador et al. had used sacB gene as the counterselection marker for screening of C. salexigens DSM 3043 mutants. However, a Ω cassette containing streptomycin resistance gene still existed in the mutant chromosome (Salvador et al. 2015). In order to make multiple gene deletion in C. salexigens, we sought to validate the suitable selection markers for unmarked allelic exchange. Plasmid pK18mobsacB (Schäfer et al. 1994) carries the pUC-based origin of replication, which makes it impossible to replicate in C. salexigens cells, and contains the sacB gene from Bacillus subtilis and a kanamycin resistance cassette. Previously, Grammann et al. had used pK18mobsacB as a suicide vector for the construction of Halomonas elongata DSM 2581 mutants impaired in biosynthesis or transport of ectoine (Grammann et al. 2002). Although C. salexigens is sensitive to kanamycin (100 μg ml−1) at low salt concentrations (≤0.6 M NaCl), we found that the minimal inhibition concentration of this antibiotic increased by four times at elevated salt concentration (2 M NaCl). The influence of salinity on the response of various moderate halophilic bacteria to the different antimicrobials was heterogeneous, generally exhibiting three different patterns (Coronado et al. 1995). The phenomena that increasing the salt concentration in growth media may lead to a reduced inhibitory effect of some antibiotics to moderate halophiles had been reported previously (Kunte and Galinski 1995; Nieto et al. 1993), although the resistance mechanism was exactly unclear. There were some evidences indicated that several antibiotics could react with ions under high salinity, which may affect its permeability across the membrane (Piekarski et al. 2009), and the chemical changes in membrane lipids in response to the high salt concentration may alter the membrane permeability toward antibiotics (Ventosa and Nieto 2005). The resistance of C. salexigens ZW4-1 toward kanamycin will result in predominantly false positives clones following conjugation. Fortunately, we found that the inhibitory effect of chloramphenicol (70 μg ml−1) toward C. salexigens was stable at 37 °C for 48 h even in the presence of 2.0 M NaCl (data not shown). Therefore, a chloramphenicol resistance cassette was PCR amplified from pACYCDuet-1 and inserted into pK18mobsacB, yielding pMDC21, which was used to select for transconjugants with plasmid integration arising from a single crossover of homologous recombination.
Generation of Δcsal_1415 mutant and phenotypic characterization of the mutant
It is well known that bacterial NhaC encodes a Na+/H+ antiporter, which plays an important role in maintaining cytoplasmic Na+ homeostasis in cellular osmoregulation (Ito et al. 1997). Eight homologous genes of nhaC were identified in the genome of C. salexigens (Ates et al. 2011). To verify the validity of above-mentioned gene replacement system, ORF csal_1415 which may code for one of the putative Na+/H+ antiporters was selected as the first gene for deletion. Plasmid pMDCΔcsal_1415 was used to introduce the mutant allele of csal_1415 lacking the internal 1116-bp DNA fragment of wild-type allele into C. salexigens ZW4-1 chromosome. Of the colonies that grew on chloramphenicol selective plates, a 507-bp fragment was amplified by colony PCR using primers sacB-F and sacB-R from all 20 colonies randomly selected, indicating that plasmid integration event had occurred (Fig. 3a). In the plasmid excision step, 100 colonies growing on sucrose-containing plates were randomly selected and transferred to D5 plates with or without chloramphenicol by toothpicks. Among them, 32% of clones showed SucrCmr phenotype, while the remaining showed SucrCms phenotype. Colony PCR with the primers sacB-F and sacB-R indicated that all SucrCms clones lost the suicide vector (Fig. 4a), while the vector backbone was still maintained in the chromosome of the SucrCmr clones, but sacB gene was inactive (data not shown). Screening for mutants among the SucrCms colonies was performed by PCR using primers 1415-uF and1415-dR, and amplifying 3006 bp from the wild type and 1890 bp from the mutant (Fig. 5a). In addition, primers 1415-test-F and 1415-test-R were used to verify that the second homologous recombination event occurred at the desired locus. The Δcsal_1415 mutant could generate the predicted 1630-bp amplicon, while the wild-type strain produces 2746-bp amplicon (Fig. 6a). In fact, none of the mutants were proved to be off-targeted, and the sequencing of PCR products also confirmed that the deletion mutants were successfully obtained. Of the 251 analyzed SucrCms colonies, 2 had the desired Δcsal_1415 genotype by PCR and DNA fragment sequencing, designated as 3043Δcsal_1415 strain.

Colony PCR analysis with primers sacB-F and sacB-R for verifying the integration of allelic exchange vector into the chromosome of C. salexigens ZW4-1 by the first homologous recombination event. a Verification of the integration of pMDCΔcsal_1415. b Verification of the integration of pMDCΔsarC. Lane M DL 5000 DNA Marker (TaKaRa); lane 1–4 chloramphenicol resistant colonies; lane P positive control: E. coli 5α (pK18mobsacB)

Colony PCR analysis with primers sacB-F and sacB-R for verifying the loss of the integrated plasmid from the chromosome of C. salexigens ZW4-1 by the second homologous recombination event. a Verifying the loss of the pMDCΔcsal_1415 backbone; b verifying the loss of the pMDCΔsarC backbone. Lane M DL 5000 DNA Marker (TaKaRa); lane 1–6 sucrose-resistant, chloramphenicol-sensitive colonies; lane P positive control: E. coli 5α (pK18mobsacB)

Verification of the deletion mutants by colony PCR. a PCR products using the primers 1415-uF and 1415-dR were separated by agarose electrophoresis resulting in a band at 3006 bp for the wild type (WT) and at 1890 bp for the Δcsal_1415 mutant (Δ). As for the additional band at approximate 1 kb, DNA sequencing results indicated that it consisted of the entire downstream homologous region of Csal_1415 due to the non-specific binding of primer 1415-uR. b PCR products using the primers sarC-uF and sarC-dR resulting in a band at 3943 bp for the wild type (WT) and at 1899 bp for the ΔsarC mutant. Lane M DL 5000 DNA marker (TaKaRa)

Verification of the allelic exchange occurred at the desired locus by colony PCR. a PCR products using the primers 1415-test-F and 1415-test-R were separated by agarose electrophoresis resulting in a band at 2746 bp for the wild type (WT) and at 1630 bp for the Δcsal_1415 mutant (Δ). b PCR products using the primers sarC-test-F and sarC-test-R resulting in a band at 4128 bp for the wild type (WT) and at 2159 bp for the ΔsarC mutant. Lane M DL 5000 DNA marker (TaKaRa)
To test the effect of csal_1415 deletion on cellular osmoadaptation, the growth of wild type and Δcsal_1415 mutant in M63 medium containing different NaCl concentrations was determined by OD600. Our results showed that the Δcsal_1415 mutant did not exhibit a salt-sensitive phenotype, and its salinity upper limit was unchanged as compared with that of wild-type strain. As shown in Fig. 7, the Δcsal_1415 mutant showed similar growth curves with those of the wild type in M63 media containing different NaCl concentrations, suggesting that ORF csal_1415 is not directly involved in osmoregulation in the presence of various NaCl concentrations, and the exactly role of this gene in C. salexigens remains to be further elucidated.

Growth characteristics of C. salexigens ZW4-1 and 3043Δcsal_1415 strains. Both strains were grown at 37 °C in M63 medium containing different NaCl concentrations with shaking. The samples were withdrawn periodically to measure the optical density at 600 nm (OD600)
Generation of ΔsarC mutant and phenotypic characterization of the mutant
To further validate the method, sarC gene deletion was performed. Based on amino acid sequence homology analysis, csal_0998, csal_0999, csal_1000 and csal_1001 code for four different subunits (β, δ, α, and γ, respectively) of heterotetrameric sarcosine oxidase (SarC), which is involved in glycine betaine catabolism and catalyzes the conversion of sarcosine to glycine (Ida et al. 2005). To introduce the deleted sarC locus into the chromosome of C. salexigens ZW4-1, plasmid pMDCΔsarC was used as the allele replacement vector. In the plasmid integration step, all of 20 randomly selected Cmr colonies contained the suicide vector integrated in the chromosomal DNA as detected by colony PCR with primers sacB-F and sacB-R (Fig. 3b). In the plasmid excision step, 36% (36/100) of Sucr colonies exhibited SucrCmr phenotype, while the remaining showed SucrCms phenotype. The SucrCms colonies were further analyzed by colony PCR with primers to check for the suicide vector (primers sacB-F and sacB-R) and mutant allele (primers sarC-uF and sarC-dR). All of the SucrCms clones tested lost the vector backbone as verified by PCR with primes sacB-F and sacB-R (Fig. 4b), and the expected in-frame deletion produced a 1.9-kb amplicon, while the wild type yielded a 3.9-kb amplicon with primers sarC-uF and sarC-dR (Fig. 5b). To further verify that the mutant allele of sarC gene was located at the desired locus, colony PCR was performed with primers sarC-test-F and sarC-test-R, resulting in a 2.1-kb fragment which is consistent with what we predicted (Fig. 6b). Of the 371 SucrCms colonies analyzed, two colonies were confirmed to be genuine mutants by PCR and DNA fragment sequencing.
To verify the biological effect of ΔsarC deletion mutation in C. salexigens, the phenotype arising from the deletion of sarC was analyzed. Since the sarcosine oxidase SarC is involved in glycine betaine catabolism, the wild type and ΔsarC mutant were tested for their abilities to utilize glycine betaine and DMG as the sole carbon source. As shown in Fig. 8, when grown in M63 minimal medium supplemented with Rif and 0.86 M NaCl, strain ZW4-1 could utilize glucose, glycine betaine and dimethylglycine as the sole carbon sources, while 3043ΔsarC strain could use glucose as the sole carbon sources, but not for glycine betaine and dimethylglycine. The phenotypes of wild-type and mutant strains are consistent with the predicted function of sarC gene in cellular metabolism. Interestingly, we found that neither wild type nor ΔsarC mutant strain could use sarcosine or glycine as the sole carbon source. The reason may appear to be lacking the appropriate transporters located on cellular membrane to transport these substrates into the cells.

Carbon utilization tests for C. salexigens wild-type and deletion mutants. a Diagram of four quadrants on a plate in which the strains were streaked. C. salexigens DSM 3043, ZW4-1, 3043ΔsarC, and 3043ΔsarCΔcsal_1415 strains were streaked on M63 agar plates containing rifampicin and 0.86 M NaCl with glucose (b), glycine betaine (c) or dimethylglycine (d) as the sole carbon source, and incubated at 37 °C for 48–60 h
Generation of Δcsal_1415ΔsarC mutant and phenotypic characterization of the mutant
One of the advantages for employing unmarked mutations is that multiple modifications can be made in a single strain using the same selection system. To test the feasibility for construction of multiple gene deletion mutant with this system in C. salexigens, Δcsal_1415ΔsarC double-gene deletion mutant was constructed. Conjugation was carried out between E. coli S17-1 λpir carrying pMDCΔcsal_1415 and C. salexigens 3043ΔsarC strain. To identify double-gene in-frame deletion mutants, colony PCR was carried out with primers the same as those used in construction of C. salexigens 3043Δcsal_1415 strain (data not shown). Among the 167 randomly selected SucrCms colonies, 2 colonies were confirmed to have the desired deletion by PCR and DNA fragment sequencing.
The growth curve in minimal medium M63 under different NaCl concentrations and the ability to utilize substrates as sole carbon source for C. salexigens 3043Δcsal_1415ΔsarC were determined under the same conditions as those for 3043Δcsal_1415 and 3043ΔsarC strains. As shown in Fig. 8, strain 3043Δcsal_1415ΔsarC could not use glycine betaine and dimethylglycine as the sole carbon source, the same as that for strain 3043ΔsarC. Based on comparison of cell growth curves, there is no significant difference among the WT, 3043Δcsal_1415, and 3043Δcsal_1415ΔsarC strains when grown in M63 medium with glucose as the sole carbon source (data not shown).
Incubation at 25 °C drastically decreases the proportion of spontaneous Sucr colonies
Although the sacB gene from B. subtilis has been used successfully as a counterselectable marker to generate unmarked mutants in many Gram-negative bacteria, one major problem using sacB gene is the occurrence of spontaneous sucrose resistance (SSR) colonies during the screening process since sacB gene was inactivated by either a mutation, or an IS element as previously described in other bacteria (Cai and Wolk 1990; Gay et al. 1985; Jager et al. 1995; van Aartsen and Rajakumar 2011), rather than lost as in authentic double recombinants. The SSR colonies still retain the integrative vector backbone in chromosome and show resistance to antibiotics, and have been observed at frequencies of 2–50% in other bacteria (Wu and Kaiser 1996). Blomfield et al. found that incubation at 30 °C on negative selection media could markedly increase sucrose sensitivity as opposed to 37 °C, and decreases the proportion of pseudo-double recombinants among the Sucr colonies (Blomfield et al. 1991). To further investigate the effect of incubation temperature on the occurrence of SSR colonies, the merodiploid in which pMDCΔsarC was integrated into the chromosome of C. salexigens ZW4-1, were inoculated in D5 medium without adding antibiotic, and incubated at 37, 30, and 25 °C, respectively. Subsequently, the cultures were individually spread on sucrose-containing plates and incubated at the same temperatures as those before. The ratios of SSR colonies among the Sucr colonies were determined to be 51 ± 9, 27 ± 10, and 1 ± 1%, respectively, indicating that growth of the merodiploid and sacB counterselection at 25 °C dramatically decreases the observed SSR rate as compared with those at 30 or 37 °C, and minimizes the number of clones to be analyzed by colony PCR.
Growth of the merodiploids in minimal medium instead of complex medium increases the recovery rate of deletion mutants
Although incubation at 25 °C in the couterselection process could significantly decrease the proportion of SSR colonies, the recovery rate of the desired mutants was only about 0.7–1.3% in C. salexigens even after the false-positive recombinants were excluded, which was not consistent with our expectation. Theoretically, if the targeted gene is not vital, the second recombination leads to either wild-type allele restoration or to allelic substitution at a 1:1 ratio when the two homologous regions flanking the target gene to be deleted were the similar sizes. The overall poor recovery of the desired mutants prompted us to test whether the presence of NaCl in the counterselection medium would affect the recovery efficiency of desired recombinants, since the presence of sodium chloride in the selective medium had been reported to be another factor that may influence the counterselection efficiency with sacB (Blomfield et al. 1991). As a moderate halophile, C. salexigens DSM 3043 requires at least 0.5 M NaCl for growth in M63 medium, and its growth can be promoted combined with other ions (O’Connor and Csonka 2003). To avoid the negative effect of NaCl on the recovery efficiency, we tried to replace NaCl with other salts in media that could support the growth of C. salexigens and found that ZW4-1 strain could grow in YET medium supplemented with MgSO4 (0.35–2.3 M) without adding NaCl. Therefore, three different kinds of media: YET medium plus 0.86 M NaCl, YET medium plus 0.42 M MgSO4, and M63 medium plus 0.86 M NaCl, were used for the growth of the merodiploids in which pMDCΔsarC was integrated into the chromosome of C. salexigens ZW4-1 in the absence of antibiotic and incubated at 25 °C with shaking for 40 h. Subsequently, the cultures were individually plated on YET agar plates containing sucrose and 0.42 M MgSO4, and incubated at 25 °C until the colonies were visible. However, our results indicated that replacement of NaCl with MgSO4 had no effect on improving the recovery efficiency of deletion mutants in C. salexigens (data not shown). Unexpectedly, the recovery rate of desired recombinants was dramatically increased in the case of growing in M63 medium plus 0.86 NaCl compared with those formerly grown in YET medium plus 0.86 M NaCl or YET medium plus 0.42 M MgSO4. Based on the result of colony PCR screening with the primers sarC-uF and sarC-dR, 32% (16/50) of SucrCms colonies formerly grown in M63 medium plus 0.86 M NaCl were verified to be genuine double crossover mutants, while only 4% (2/50) and 2% (1/50) of colonies formerly grown in YET medium with NaCl and YET medium with MgSO4 carried the sarC deletion, which indicated that the medium composition, not just confined to NaCl, had an obvious influence on the recovery rate of deletion mutants, although the exact reasons for this phenomenon remain unknown. To further investigate the role of sucrose-containing media on the recovery rate of the desired recombinants, the culture formerly grown in M63 medium plus 0.86 M NaCl was plated on M63 plates containing 0.86 M NaCl and sucrose, D5 plates containing sucrose, and YET plates containing 0.42 M MgSO4 and sucrose, respectively, but there was no obvious effect on increasing the recovery rate of desired recombinants (data not shown). Since ZW4-1 strain reached the highest growth rate when cultured in D5 medium, D5 medium supplemented with sucrose was ultimately employed as the negative selection medium.
Conclusions
In this report, a markerless gene deletion system via allelic exchange was first successfully developed in C. salexigens, which can be used for functional gene analysis as well as for other applications, such as multiple point mutations or insertion in C. salexigens chromosome DNA, and it expands the genetic tools for Chromohalobacter. Critically, our results show that growth of the merodiploids and sucrose screening at 25 °C is more effective to decrease the occurrence of spontaneous sucrose resistance colonies than at higher temperature with sacB gene as the couterselection marker in C. salexigens, and growth of the merodiploids in mineral salt medium instead of complex medium is critical to dramatically increase the recovery rate of deletion mutants. These new findings not only are applicable for the construction of deletion mutants in C. salexigens, but also may shed light on allelic exchange with sacB gene as the counterselectable marker in the other halophiles or marine microorganisms, which need the necessary presence of NaCl for the normal growth.
References
Arahal DR, Garcia MT, Vargas C, Canovas D, Nieto JJ, Ventosa A (2001) Chromohalobacter salexigens sp. nov., a moderately halophilic species that includes Halomonas elongata DSM 3043 and ATCC 33174. Int J Syst Evol Microbiol 51:1457–1462
Argandoña M, Vargas C, Reina-Bueno M, Rodríguez-Moya J, Salvador M, Nieto JJ (2012) An extended suite of genetic tools for use in bacteria of the Halomonadaceae: an overview. In: Lorence A (ed) Recombinant gene expression. Humana Press, Totowa, pp 167–201
Ates O, Oner ET, Arga KY (2011) Genome-scale reconstruction of metabolic network for a halophilic extremophile, Chromohalobacter salexigens DSM 3043. BMC Syst Biol 5:12
Blomfield IC, Vaughn V, Rest RF, Eisenstein BI (1991) Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol Microbiol 5:1447–1457
Bortesi L, Zhu C, Zischewski J, Perez L, Bassié L, Nadi R, Forni G, Lade SB, Soto E, Jin X, Medina V, Villorbina G (2016) Patterns of CRISPR/Cas9 activity in plants, animals and microbes. Plant Biotechnol J 14:2203–2216
Cai YP, Wolk CP (1990) Use of a conditionally lethal gene in Anabaena sp. strain PCC 7120 to select for double recombinants and to entrap insertion sequences. J Bacteriol 172:3138–3145
Canovas D, Vargas C, Csonka LN, Ventosa A, Nieto JJ (1996) Osmoprotectants in Halomonas elongata: high-affinity betaine transport system and choline-betaine pathway. J Bacteriol 178:7221–7226
Canovas D, Vargas C, Iglesias-Guerra F, Csonka LN, Rhodes D, Ventosa A, Nieto JJ (1997) Isolation and characterization of salt-sensitive mutants of the moderate halophile Halomonas elongata and cloning of the ectoine synthesis genes. J Biol Chem 272:25794–25801
Copeland A, O’Connor K, Lucas S, Lapidus A, Berry KW, Detter JC, Del Rio TG, Hammon N, Dalin E, Tice H, Pitluck S, Bruce D, Goodwin L, Han C, Tapia R, Saunders E, Schmutz J, Brettin T, Larimer F, Land M, Hauser L, Vargas C, Nieto JJ, Kyrpides NC, Ivanova N, Goker M, Klenk HP, Csonka LN, Woyke T (2011) Complete genome sequence of the halophilic and highly halotolerant Chromohalobacter salexigens type strain (1H11T). Stand Genomic Sci 5:379–388
Coronado MJ, Vargas C, Kunte HJ, Galinski EA, Ventosa A, Nieto JJ (1995) Influence of salt concentration on the susceptibility of moderately halophilic bacteria to antimicrobials and its potential use for genetic transfer studies. Curr Microbiol 31:365–371
García-Estepa R, Argandoña M, Reina-Bueno M, Capote N, Iglesias-Guerra F, Nieto JJ, Vargas C (2006) The ectD gene, which is involved in the synthesis of the compatible solute hydroxyectoine, is essential for thermoprotection of the halophilic bacterium Chromohalobacter salexigens. J Bacteriol 188:3774–3784
Gay P, Le Coq D, Steinmetz M, Ferrari E, Hoch JA (1983) Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J Bacteriol 153:1424–1431
Gay P, Le Coq D, Steinmetz M, Berkelman T, Kado CI (1985) Positive selection procedure for entrapment of insertion sequence elements in Gram-negative bacteria. J Bacteriol 164:918–921
Grammann K, Volke A, Kunte HJ (2002) New type of osmoregulated solute transporter identified in halophilic members of the bacteria domain: TRAP transporter TeaABC mediates uptake of ectoine and hydroxyectoine in Halomonas elongata DSM 2581T. J Bacteriol 184:3078–3085
Horton RM (1997) In vitro recombination and mutagenesis of DNA. SOEing together tailor-made genes. Methods Mol Biol 67:141–149
Ida K, Moriguchi T, Suzuki H (2005) Crystal structure of heterotetrameric sarcosine oxidase from Corynebacterium sp. U-96. Biochem Biophys Res Commun 333:359–366
Ito M, Guffanti AA, Zemsky J, Ivey DM, Krulwich TA (1997) Role of the nhaC-encoded Na+/H+ antiporter of alkaliphilic Bacillus firmus OF4. J Bacteriol 179:3851–3857
Jager W, Schafer A, Kalinowski J, Puhler A (1995) Isolation of insertion elements from Gram-positive Brevibacterium, Corynebacterium and Rhodococcus strains using the Bacillus subtilis sacB gene as a positive selection marker. FEMS Microbiol Lett 126:1–6
Kunte HJ, Galinski EA (1995) Transposon mutagenesis in halophilic eubacteria: conjugal transfer and insertion of transposon Tn5 and Tn1732 in Halomonas elongata. FEMS Microbiol Lett 128:293–299
Manilla-Pérez E, Lange AB, Hetzler S, Wältermann M, Kalscheuer R, Steinbüchel A (2010) Isolation and characterization of a mutant of the marine bacterium Alcanivorax borkumensis SK2 defective in lipid biosynthesis. Appl Environ Microbiol 76:2884–2894
Nieto JJ, Fernandez-Castillo R, Garcia MT, Mellado E, Ventosa A (1993) Survey of antimicrobial susceptibility of moderately halophilic eubacteria and extremely halophilic aerobic archaeobacteria: utilization of antimicrobial resistance as a genetic marker. Syst Appl Microbiol 16:352–360
O’Connor K, Csonka LN (2003) The high salt requirement of the moderate halophile Chromohalobacter salexigens DSM 3043 can be met not only by NaCl but by other ions. Appl Environ Microbiol 69:6334–6336
Piekarski T, Buchholz I, Drepper T, Schobert M, Wagner-Doebler I, Tielen P, Jahn D (2009) Genetic tools for the investigation of Roseobacter clade bacteria. BMC Microbiol 9:265
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Csa9 system. Nat Protoc 8:2281–2308
Reina-Bueno M, Argandoña M, Salvador M, Rodríguez-Moya J, Iglesias-Guerra F, Csonka LN, Nieto JJ, Vargas C (2012) Role of trehalose in salinity and temperature tolerance in the model halophilic bacterium Chromohalobacter salexigens. PLoS One 7:e33587
Reyrat JM, Pelicic V, Gicquel B, Rappuoli R (1998) Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect Immun 66:4011–4017
Rodríguez-Moya J, Argandoña M, Reina-Bueno M, Nieto JJ, Iglesias-Guerra F, Jebbar M, Vargas C (2010) Involvement of EupR, a response regulator of the NarL/FixJ family, in the control of the uptake of the compatible solutes ectoines by the halophilic bacterium Chromohalobacter salexigens. BMC Microbiol 10:256
Salvador M, Argandoña M, Pastor JM, Bernal V, Cánovas M, Csonka LN, Nieto JJ, Vargas C (2015) Contribution of RpoS to metabolic efficiency and ectoines synthesis during the osmo- and heat-stress response in the halophilic bacterium Chromohalobacter salexigens. Environ Microbiol Rep 7:301–311
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73
Simon R, Priefer U, Pühler A (1983) A broad host-range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Nat Biotechnol 1:784–791
Tokunaga H, Mitsuo K, Ichinose S, Omori A, Ventosa A, Nakae T, Tokunaga M (2004) Salt-inducible multidrug efflux pump protein in the moderately halophilic bacterium Chromohalobacter sp. Appl Environ Microbiol 70:4424–4431
van Aartsen JJ, Rajakumar K (2011) An optimized method for suicide vector-based allelic exchange in Klebsiella pneumoniae. J Microbiol Methods 86:313–319
Vargas C, Nieto JJ (2004) Genetic tools for the manipulation of moderately halophilic bacteria of the family Halomonadaceae. Methods Mol Biol 267:183–208
Vargas C, Argandoña M, Reina-Bueno M, Rodríguez-Moya J, Fernández-Aunión C, Nieto JJ (2008) Unravelling the adaptation responses to osmotic and temperature stress in Chromohalobacter salexigens, a bacterium with broad salinity tolerance. Saline Syst 4:14
Ventosa A, Nieto JJ (1995) Biotechnological applications and potentialities of halophilic microorganisms. World J Microbiol Biotechnol 11:85–94
Ventosa A, Nieto JJ (2005) Contribution of chemical changes in membrane lipids to the osmoadaptation of the halophilic bacterium Chromohalobacter salexigens. Syst Appl Microbiol 28:571–581
Vreeland RH, Litchfield CD, Martin EL, Elliot E (1980) Halomonas elongata, a new genus and species of extremely salt-tolerant bacteria. Int J Syst Evol Microbiol 30:485–495
Wu SS, Kaiser D (1996) Markerless deletions of pil genes in Myxococcus xanthus generated by counterselection with the Bacillus subtilis sacB gene. J Bacteriol 178:5817–5821
Yin J, Chen JC, Wu Q, Chen GQ (2015) Halophiles, coming stars for industrial biotechnology. Biotechnol Adv 33:1433–1442
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Number 31070047). We are grateful to Professor Alexander Steinbüchel (Institut für Molekulare Mikrobiologie und Biotechnologie, Westfälische Wilhelms-Universität Münster, Germany) for kindly providing E. coli SM10 (λpir) and pUTmini-Tn5Cm, and Professor Song Yang (Qingdao Agricultural University) for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by L. Huang.
Rights and permissions
About this article

Cite this article
Shao, YH., Guo, LZ., Yu, H. et al. Establishment of a markerless gene deletion system in Chromohalobacter salexigens DSM 3043. Extremophiles 21, 839–850 (2017). https://doi.org/10.1007/s00792-017-0946-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-017-0946-y