
Abstract
A pullulanase-encoding gene from psychrotrophic Exiguobacterium sp. SH3 was cloned and expressed in both E. coli and Bacillus subtilis. The functional recombinant enzyme (Pul-SH3) was purified as a His-tagged protein. Pul-SH3 was characterized to be a cold-adapted type I pullulanase with maximum activity at 45 °C. Using fluorescence spectroscopy, the melting temperature of Pul-SH3 was determined to be about 52 °C. The enzyme was able to hydrolyze pullulan, soluble starch, potato starch, and rice flour. It was exceptionally tolerant in the pH range of 4–11, exhibiting maximum activity at pH 8.5 and more than 60 % of the activity in the pH range of 5–11. Being a detergent-tolerant pullulanase, Pul-SH3 retained 99, 89, and 54 % of its activity at 10 % concentration of Triton-X100, Tween 20, and SDS, respectively. The enzyme also exhibited an activity of 80.4 and 93.7 % in the presence of two commercial detergents, Rika (7.5 % v/v) and Fadisheh (2.5 % w/v), respectively. The enzyme was even able to remain active by 54.5 and 85 % after 10-day holding with the commercial detergents. Thermal stability of the enzyme could w on silica. Pul-SH3 with several industrially important characteristics seems desirable for cold hydrolysis of starch.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pullulanase, α-dextrin 6-glucanohydrolase (EC 3.2.1.41), is a glycosidase with the ability to hydrolyze α-1,6-glycosidic linkages in pullulan and starch. The enzyme has been purified from various microbial sources (Kar et al. 2012; Plant et al. 1987; Qoura et al. 2014; Saha et al. 1988). In recent years, many attempts have been made for heterologous expression of pullulanase-encoding genes and characterization of the recombinant proteins (Kang et al. 2011; Li et al. 2012; Qiao et al. 2015; Wu et al. 2014). Based on substrate specificity, pullulanase enzymes are classified into type I and II. The type I pullulanase is known to specifically hydrolyze α-1,6 linkages in pullulan and starch. In contrast, pullulanase type II (also known as amylopullulanase) has been found to hydrolyze α-1,6 linkages in pullulan as well as both α-1,6 and α-1,4 linkages in starch. Pullulan hydrolases are another group of pullulan-hydrolyzing enzymes that are in turn categorized into types I, II, and III. Pullulan hydrolase types I and II break the α-1,4 linkages in pullulan to release panose and isopanose, respectively, while pullulan hydrolase type III breaks both α-1,4 and α-1,6 linkages to release maltotriose, maltose, panose and glucose (Hii et al. 2012).
Starch is regarded as the second most abundant biological matter after cellulose (Corre et al. 2010). This natural polymer has widely been used by humans in food, pharmaceutical, paper, packaging, adhesive, and textile industries. Starch is an invaluable biomass which, besides the traditional applications, has recently been used as a replacement of petroleum in fuel and commodity manufacturing. The consumption of petroleum-based fuels and products has resulted in serious environmental concerns over the accumulation of greenhouse gases and intractable wastes. Therefore, there has been a growing tendency in recent years to use biomass-based fuel and commodities rather than the petroleum-based ones (Clark et al. 2006). In this context, starch has increasingly been used for production of fuel and recyclable utensils including dishes and cups. Starch-active enzymes already account for a large portion of the world enzyme market and there seems to be a growing demand for the enzymes in novel applications (Nguyen et al. 2002). Pullulanase as a starch-debranching enzyme has gained attention due to its potential application in starch-related industries such as starch processing, laundry, waste recycling, and biofuel production. It has been shown that the application of pullulanase along with amylases makes a significant difference in the rate of starch hydrolysis and product formation (Kennedy et al. 1988; Roy and Gupta 2004). In particular, pullulanase type I, by specific degradation of α-1,6 glucosidic linkages, is able to break down starch to polymeric chains of α-1,4-linked glucose units. The chains may subsequently be hydrolyzed by an appropriate glucoamylase or amylase to produce glucose or maltose (Bertoldo et al. 1999; Sapińska et al. 2014). In addition, the processing of starch granules at high temperatures results in intermolecular cross-linking and increased viscosity of starch slurry (Chung et al. 2002). The high viscosity is a serious problem in the high-concentration starch hydrolysis that can be resolved by pullulanase type I (Hong et al. 2015). Current technologies are mostly based on starch hydrolysis at temperatures above 60 °C and, therefore, a thermotolerant type I pullulanase seems suitable for industrial applications (Bertoldo et al. 1999). On the other hand, new technologies are developing to function at ambient temperature (Cinelli et al. 2015). Starch hydrolysis without heating is deemed to be superior in terms of costs and the overall yield. Almost all of the pullulanases reported so far are mesophilic and thermophilic enzymes, while cold-adapted pullulanase has been brought into attention by just a few recent studies (Elleuche et al. 2015; Rajaei et al. 2014). We have previously isolated and characterized Exiguobacterium sp. SH3 as a pullulanase-producing psychrotrophic bacterium (Rajaei et al. 2014). In this manuscript, we report on the cloning of a pullulanase-encoding gene from this bacterium and the heterologous expression of the gene in both E. coli and Bacillus subtilis. The pullulanase (Pul-SH3) was purified to homogeneity and characterized using standard biochemical procedures.
Materials and methods
Bacterial strains and gene cloning
Exiguobacterium sp. SH3 was isolated in a previous study (Mojallali et al. 2014). The gene encoding for the pullulanase type I was identified in this bacterium from a partial genomic library that was previously constructed (Emampour et al. 2015). The gene was amplified by the PCR technique using genomic DNA of the bacterium as template and the following specific primers: 5′-ATA GAA TTC AGG AGG ATT AAC TAA TGA ATC GAT TGA AAT CAG TCT GTG C-3′ and 5′-ATA CTC GAG TTT TTT TAA CAC GTA AGA ACT CAG TGG G-3′ containing the EcoRI and XhoI restriction sites, respectively. The PCR product was inserted into the expression vector pET-26b(+) (Novagen), and the resulting plasmid, pET-pul, was used for the transformation of E. coli BL21 (DE3) using kanamycin (50 µg ml−1) as selection marker. Besides, a pair of primers, including 5′- ATA GAA TTC AGG AGG ATT AAC TAA TGA ATC GAT TGA AAT CAG TCT GTG C-3′ and 5′- ATA TCT AGA TTA GTG GTG GTG GTG GTG GTG TTT TTT TAA CAC GTA AGA ACT CAG TGG G-3′, was designed to insert the gene into the shuttle vector pHY300PLK (Takara Bio, Shiga, Japan). In the forward primer, the restriction sequence of EcoRI and in the reverse primer the restriction sequence of XbaI along with a sequence encoding for 6× His were included. E. coli DH5α was used as a cloning host and the transformants were selected on LB medium containing ampicillin (30 µg ml−1). The resulting plasmid, pHY-pul, was confirmed by sequencing and used for the transformation of B. subtilis WB600 as the expression host. The B. subtilis WB600 transformants were selected on LB medium containing tetracycline (20 µg ml−1).
Gene expression and enzyme purification
An overnight culture of the E. coli BL21 (DE3) cells harboring pET-pul was used for inoculation of 20 ml of fresh LB medium to an optical density (600 nm) of 0.1 in a 100 ml flask. The cells were grown in a shaking incubator at 37 °C and 180 rpm to an OD of about 1.5 (600 nm). The cells were then induced by 1 mM IPTG (isopropyl-1-thio-β-d-galactoside) and allowed to grow for 24 h at 22 °C and 120 rpm before being harvested by a 15-min centrifugation (5000g, 4 °C). The resulting pellet was resuspended in the lysis buffer [50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 20 mM 2-ME (β-mercaptoethanol), 1 mM PMSF (phenylmethanesulfonyl fluoride), pH 8.0] and sonicated on ice at 90 % amplitude and 0.7 s cycles for 6 times each 45 s with 45 s intervals (Hielscher Ultrasonics 108 GmbH, Teltow, Germany). After centrifugation (10,000g, 4 °C, 30 min), 2 ml of the cell-free supernatant with pullulanase activity was added to 1 ml of equilibrated Ni–NTA (nickel–nitrilotriacetic acid) agarose and mixed gently on a rotary shaker for 60 min. The mixture was loaded into a column, which was then washed with 10 ml of washing buffer I [50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole (pH 8.0)]. Next, the column was washed with 10 ml of washing buffer II [washing buffer I with 1 % Triton X-100, 1 % Tween 20, 10 mM 2-ME, 2 M NaCl and 10 % glycerol], and again with washing buffer I until no protein was detected spectrophotometrically (at 280 nm) in the flow-through. Finally, the column was washed with 4 ml of elution buffer [50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole (pH 8.0)], while the eluate was collected in 1 ml fractions. The fractions were analyzed for activity and purity using pullulanase assay and SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis), respectively. Fractions containing the purified pullulanase (Pul-SH3) were pulled together and dialyzed overnight against Tris–HCl buffer (pH 8.5). In the case of B. subtilis WB600, the transformed cells harboring pHY-pul were grown on the tetracycline-containing LB broth at 37 °C for 24 h. The culture medium was then centrifuged and the cell-free supernatant containing Pul-SH3 was used for protein purification in the same way as described above with the exception that the washing step with buffer II was eliminated.
SDS-PAGE and zymography
SDS-PAGE was conducted using 2 % stacking and 10 % separating gels to determine the purity of Pul-SH3. Activity staining was done in the same way as SDS-PAGE with the following modifications: 1 % red-pullulan was added to the resolving gel and after electrophoresis the gel was soaked in Tris–HCl buffer (pH 8.5) containing 1 % Triton X-100 for 30 min at 4 °C. Next, the gel was incubated in Tris–HCl buffer (pH 8.5) at 40 °C until a clear band resulting from in-gel hydrolysis of red-pullulan appeared. Protein determination at each step was performed according to the Bradford procedure (Bradford 1976).
Activity assay
Pullulanase activity was assayed by the DNS (3,5-dinitrosalicylic acid) method according to Miller (1959) with the following modifications. The reaction mixture was composed of 150 µL of 1 % pullulan prepared in 200 mM Tris–HCl buffer (pH 8.5) and 150 µL of the purified enzyme. After 30 min incubation at 45 °C, the reaction was stopped by adding 900 µL of DNS, followed by 5 min heating in boiling water and cooling on ice. The blank sample was used to normalize the non-enzymatic release of reducing sugars. The amount of reducing sugars was measured spectrophotometrically at 540 nm using glucose as standard. One unit of enzyme activity was defined as the amount of enzyme required to release 1 µmol of glucose equivalent of reducing sugar per minute.
Temperature and pH profile of activity
The influence of temperature on the activity of Pul-SH3 was investigated by conducting the enzyme reaction at different temperatures in the range of 0–80 °C. The thermal stability was analyzed by holding the enzyme at different temperatures including 30, 40, 50, 60, and 70 °C for 90 min. The residual activity of the enzyme was measured at the optimum activity temperature after 5, 15, 30, 60, and 90 min holding at each temperature.
The activity of Pul-SH3 as a function of pH was investigated in the range of pH 4–11 using citrate buffer (pH 4–6), phosphate buffer (pH 6–7), Tris HCl buffer (7–9), and Gly-NaOH buffer (9–11).
Enzyme immobilization on silica particles
The epoxy-functionalized silica was kindly provided by Dr. Mohammadi (Ashjari et al. 2015). Immobilization was carried out via a simple adsorption technique using purified enzyme dialyzed against phosphate buffer (pH 7.0). For this purpose, 80 mg of the silica particles was added to 2 ml of the enzyme solution (0.4 mg ml−1). The mixture was gently stirred at room temperature for 16 h. At the end, the immobilized enzyme was separated from the solution by centrifugation (8000g, 30 min). The yield of the enzyme immobilization was monitored by spectrometric determination (280 nm) of the remaining enzyme in solution. The thermal stability of the immobilized enzyme was assayed by holding at various temperatures in the range of 30–70 °C with 10 °C increments. The residual activity of the immobilized enzyme was measured after 5, 15, 30, 60, and 90 min holding at each temperature.
Fluorescence spectroscopy
The thermal unfolding of Pul-SH3 was monitored by fluorescence spectroscopy. Fluorescence emission was analyzed at various temperatures ranging from 10 to 80 °C with 10 °C increments in a Cary-Eclipse spectrofluorimeter (Varian, Australia). The purified enzyme (50 µg ml−1) was excited at 280 nm and fluorescence spectrum for each temperature was recorded at 300–500 nm. All spectra were corrected for the buffer contribution and the barycentric mean fluorescence (BCM) was determined according to the following equation:
where I(λ) is the fluorescence intensity at wavelength λ.
The melting temperature (T m) was calculated using barycentric fluorescence as a function of temperature according to the following relation:
where max is the local maximum and dBCM/dT(T) is the first derivative of barycentric fluorescence as a function of temperature (Garstka et al. 2015).
Effect of chemical modulators and cations
To study the impact of various chemicals on the activity of Pul-SH3, enzyme assays were conducted in the presence of metal ions, sugars, denaturants, and detergents. Metal ions were included in the reaction mixture at 2 mM final concentration. The impact of sugars including manitol, sorbitol, glucose, maltose, myoinositol, sucrose, and glycerol was studied at 0.2 % final concentration. Denaturants and reducing agents were used at the following concentrations: 2-ME, 1 %; GuHCl (guanidine hydrochloride), 0.2 and 1 %; DTT (dithiothreitol), 0.2 %; urea, 1 and 10 %; (NH4)2SO4, 0.2 and 1 %; iodoacetamide, 0.2 %; and EDTA (ethylenediaminetetraacetic acid), 1 and 10 mM. Non-ionic detergents including Triton X-100, and Tween 20 were used at 10 % concentration. The SDS (sodium dodecyl sulfate) as an anionic detergent was used at both 1 and 10 % concentrations.
Substrate specificity
The substrate specificity of Pul-SH3 was analyzed using pullulan, soluble starch, potato starch, amylopectin, maltodextrin, amylose, and dextran (all from Sigma-Aldrich) as well as rice flour and wheat flour (purchased from a local bakery). The enzyme activity on each substrate was assayed under the standard conditions as described above.
Thin-layer chromatography (TLC)
The hydrolysis products of Pul-SH3 were analyzed by TLC. For this purpose, the enzyme reactions were conducted under the optimum temperature and pH conditions using pullulan and starch as substrates. An aliquot of 5 µL from each reaction and 1 µL of a standard marker containing glucose, maltose, maltotriose, maltotetraose, maltopentaose, maltohexaose, and maltoheptaose (5 mg ml−1, Sigma-Aldrich) were separately blotted onto a silica gel plate (60 F254 plastic sheet, 20–20 cm, Merck, Germany). The TLC was run using a solvent system composed of n-butanol:ethanol:water (5:5:3 v/v). At the end, the TLC plate was left to dry at room temperature, sprayed with a methanol:sulfuric acid (9:1 v/v) solution, and heated for 10 min at 110 °C to visualize the sugar spots.
Results
Bioinformatic analysis
The pullulanase-encoding gene (Pul-SH3) was identified in the genome of Exiguobacterium sp. SH3 as an open reading frame of 2913 nucleotide base pairs. The translated amino acid sequence of the gene was analyzed using bioinformatic tools to reveal the tructural characteristics of the putative pullulanase. An analysis conducted by SignalP server (http://www.cbs.dtu.dk/services/SignalP) indicated that the enzyme contained a signal peptide of 27 amino acids. This signal peptide was predicted to be similar to that of Gram-positive bacteria. A conserved-domain analysis using the CDD database (http://www.ncbi.nlm.nih.gov/cdd) indicated that Pul-SH3 differs from other type I pullulanases in terms of modular structure (Fig. 1a). The primary structure of Pul-SH3 contains two family 41 carbohydrate-binding modules (CBM41) at the N-terminal end, followed by an E-set domain, and then a (β/α)8 TIM-barrel domain of family 13 of the glycosyl hydrolases toward the C-terminus. The E-set domains (Conserved Domain Database: cl09101) may be related to the immunoglobulin and/or fibronectin type III superfamilies. According to the CDD database, a subset of E-set domains, found in carbohydrate-active enzymes such as pullulanase, belongs to the family 48 carbohydrate-binding module (CBM48).

a A comparison of the modular structure between Pul SH3 from Exiguobacterium sp. SH3 and several type I pullulanses from other bacteria. CBM41 and CBM48 are carbohydrate-binding modules of family 41 and 48; E-set is the early set domain. b The conserved regions of type I pullulanases. Conserved regions (I–IV) and the signature sequence (SS-Pul I) of the type I pullulanases are highlighted. The catalytic residues (Asp-Glu-Asp) are shown by asterisks
One of the remarkable features of Pul-SH3 is the low amino acid similarity with characterized type I pullulanases from other bacteria (Table 1). However, Pul-SH3 contains all the conserved regions of a type I pullulanase as revealed by the amino acid alignment (Fig. 1b). Accordingly, the putative catalytic triad of Pul-SH3 was identified to be composed of Asp666, Glu695, and Asp786. The phylogenetic relation of Pul-SH3 with other type I pullulanases is depicted in Fig. 2.

Phylogenetic relation of Pul-SH3 with several other type I pullulanases. The tree was constructed by the neighbor-joining method using MEGA6 software. The numbers represent bootstrap values (%) based on 1000 replicates. The scale bar represents 0.2 substitutions per amino acid position
Gene cloning and expression
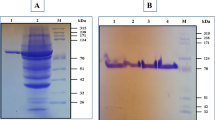
Based on the nucleotide sequence of Pul-SH3 retrieved from a partial genome sequence of Exiguobacterium sp.SH3 (Emampour et al. 2015), a pair of primers were designed and used for PCR amplification of a 2913-bp pullulanase-encoding gene from the bacterium. The gene was inserted into pET-26b(+) and cloned in E. coli DH5α. The resulting plasmid, pET-pul, was confirmed by sequencing and further was used for the gene expression in E. coli BL21(DE3). The gene was also inserted into pHY300PLK to construct pHY-pul plasmid to be used for the gene expression in B. subtilis. The expression of the gene in E. coli BL21 (DE3) under the control of T7 promoter using 1 mM IPTG resulted in cytoplasmic accumulation of the pullulanase. The pullulanase was purified as a C-terminal His-tagged protein using metal affinity chromatography. The SDS-PAGE of the purified protein with a calculated molecular mass of 110 kDa is shown in Fig. 3. The activity of the purified protein was confirmed by zymography. In contrast, the gene expression in B. subtilis WB600 using recombinant pHY-pul plasmid was accompanied with the secretion of the enzyme to the culture medium. This observation was in agreement with the prediction of a signal peptide in the structure of Pul-SH3. However, there was not any significant difference in activity observed between recombinant enzymes purified from E. coli BL21 (DE3) and B. subtilis WB600.

SDS-PAGE and zymogram analysis of Pul-SH3. 1 Cell lysate of induced E. coli BL21 (DE3) harboring pET-26b(+); 2 cell lysate of induced E. coli BL21 (DE3) harboring pET-pul; 3–5 fractions of purified protein from E. coli BL21 (DE3); 6,7 culture supernatant of induced B. subtilis WB600 harboring pHY-pul; 8–11 fractions of purified protein from B. subtilis WB600; M-marker; Z-functional activity of purified enzyme as shown on a zymogram
Substrate specificity
The substrate specificity of Pul-SH3 was analyzed using pullulan, amylopectin, soluble starch, potato starch, dextran, maltodextrin, wheat flour, rice flour, and amylose. As shown in Table 2, the maximum activity of the enzyme was obtained on pullulan. The enzyme was also able to hydrolyze soluble starch, rice flour, potato starch, and amylopectin. The enzyme activity on wheat flour and maltodextrin was negligible; no activity was detected on amylose as is common among type I pullulanases. The TLC analysis revealed that the hydrolysis of pullulan by Pul-SH3 resulted in the release of maltotriose, while the starch hydrolysis resulted in the formation of a mixture of maltose, maltotriose, and maltotetraose (Fig. 4).

Thin-layer chromatography (TLC) of hydrolysis products of Pul-SH3 from starch (lane 1) and pullulan (lane 2). A standard mixture (lane M), containing glucose (G1), maltose (G2), maltotriose (G3), maltotetraose (G4), maltopentaose (G5), maltohexaose (G6), and maltoheptaose (G7) was used as a molecular marker
Impact of temperature and pH on the enzyme activity
The impact of temperature on the pullulan- and starch-hydrolysis activity of Pul-SH3 was studied in the range of 0–80 °C. Activity assays were conducted at each temperature under the standard conditions. The results showed that the enzyme was active at all tested temperatures with maximum activity at 45 and 40 °C for pullulan and starch hydrolysis, respectively (Fig. 5a, b). The temperature profile of the pullulanase activity was almost symmetrical, illustrating a relative activity above 30 % of the maximum value in the range of 10–70 °C. However, the enzyme was most active in the temperature range of 40–50 °C. The analysis of thermal stability revealed that Pul-SH3 was stable at 30 °C, but at higher temperatures the enzyme was prone to instability. The loss of activity at 40 °C was minor, and after 90 min holding at the temperature the residual activity was still about 92 %. However, the loss of activity at higher temperatures was proportionally accelerated, so that after 5 min holding at 50 °C the residual activity dropped to 83 %, and at 60 and 70 °C the enzyme was totally inactivated (Fig. 5c).

The impact of temperature on the activity and stability of Pul-SH3. Temperature profile of Pul-SH3 activity on pullulan (a) and starch (b) was analyzed under standard conditions at various temperatures. Thermal stability of Pul-SH3 was studied by holding the enzyme for 90 min at 30 °C (filled circle), 40 °C (fillled square), 50 °C (filled diamond), 60 °C (filled triangle), and 70 °C (filled inverted triangle). The residual activity was analyzed at intervals under standard conditions at 45 °C using pullulan as substrate (c). Thermal stability of immobilized enzyme on silica support was studied in the same way as done with free enzyme (d)
However, the immobilization of Pul-SH3 on silica support significantly improved the thermal stability, so that the immobilized enzyme was quite stable during a 90-min holding at 50 °C. The immobilization also improved the thermal stability at 60 and 70 °C, so that the enzyme was able to retain about 53 and 21 % of the maximum activity after 30-min holding at the temperatures, respectively (Fig. 5d).
The impact of temperature on the structure of Pul-SH3 was studied by intrinsic fluorescence spectroscopy. The continued decrease of fluorescence intensity with temperature in the range of 10–80 °C may be explained as an indication to the conformational modifications related to unfolding and misfolding of the protein (Fig. 6a). The fluorescence spectra showed that the solvent exposure of tryptophan residues and the overall folding status of Pul-SH3 tend to alter with temperature. The melting temperature of the enzyme was accordingly calculated to be about 52 °C (Fig. 6b).

Thermal analysis of Pul-SH3 unfolding using fluorescence spectroscopy. a Fluorescence spectra were obtained at various temperatures using 50 µg ml−1 of purified enzyme. b Thermal denaturation of Pul-SH3 expressed as barycentric mean fluorescence (BCM). The graph fitted to first derivate of BCM shows melting temperature, Tm, as a peak maximum (dashed)
The impact of pH on the activity of Pul-SH3 was analyzed in the range of pH 4–11 (Fig. 7). Interestingly, the results showed that Pul-SH3 with an optimum pH of 8.5 was active at all tested pH points. More interestingly, the enzyme was able to exhibit high activity (above 60 %) over a broad range of pH (5–11) (Fig. 7a). Also, the enzyme was able to retain its starch-hydrolysis activity over the entire pH range with maximum activity at pH 7 (Fig. 7b).

The impact of pH on pullulan- and starch-hydrolysis activity of Pul-SH3. Reactions were conducted using purified enzyme under standard conditions in citrate buffer (filled circle), phosphate buffer (filled triangle), Tris–HCl buffer (fillled square), and Gly–NaOH buffer (filled diamond) at 45 °C for pullulanase activity (a), and at 40 °C for starch-hydrolysis activity (b)
Impact of various chemicals on enzyme activity
Reducing agents including 2-ME and DTT stimulated the activity of Pul-SH3 by about 50 % (Table 3). Pul-SH3 exhibited remarkable stability against denaturants including GuHCl (1 %), urea (10 %), ammonium sulfate (1 %), and iodoacetamide (0.2 %). Among the sugars, just maltose showed an inhibitory effect resulting in 25 % decrease in enzyme activity. Other sugars neither stimulated nor inhibited the activity. Pul-SH3 was quite stable against detergents, so that the enzyme activity was not affected by Triton X-100 and was only slightly decreased by Tween 20 at 10 % concentration of the non-ionic detergents. Besides, SDS as a strong denaturing agent did not affect the enzyme activity at 1 % concentration. Remarkably, the enzyme even in the presence of high concentration of SDS (10 %) was able to retain about 54 % of its activity. The enzyme activity was also not affected by EDTA, indicating that Pul-SH3 is a metal-independent pullulanase. Among the cations tested, Mn2+ significantly stimulated the activity by 40 %, while Zn+2 and Cu2+ inhibited the enzyme activity by 52 and 22 %, respectively. Other cations did not alter the enzyme activity (Table 3).
Given the remarkable activity of Pul-SH3 in the presence of SDS, two commercial detergents, Rika (7.5 % v/v) and Fadisheh (2.5 % w/v), were used to assess the potential application of the enzyme for washing purposes. The results showed that the enzyme was highly active in the presence of the detergents by 80.4 and 93.7 %, respectively. In addition, the stability of the enzyme against the commercial detergents was interestingly high, so that the remaining activity after a 10-day holding at room temperature with Rika (7.5 % v/v) and Fadisheh (2.5 % w/v) was about 54.5 and 85 %, respectively.
Determination of kinetic parameters
The maximum velocity (V max) and the Michaelis constant (K m) of Pul-SH3 were estimated using various concentrations of pullulan in the range of 0.3–40 mg ml−1. Assays were conducted in three independent experiments under standard conditions and the resulting data were analyzed using SigmaPlot software (Systat, San Jose, CA, USA). The V max and K m values of the enzyme were estimated to be about 20.3 U mg−1 and 2.8 mg ml−1, respectively. The catalytic constant (K cat) and catalytic efficiency (K cat/ K m) were calculated to be 37 s−1 and 13.2 ml mg−1 s−1, respectively.
Discussion
Although many articles have been published on different pullulanases, just few have addressed the characterization of type I pullulanase and only one has been specifically on the cold-adapted type I pullulanase (Elleuche et al. 2015). Type I pullulanase has been suggested as an industrial enzyme, in combination with amylases, for starch processing and sugar syrup production (Roy and Gupta 2004; Bertoldo et al. 1999). In the current study, the gene encoding for Pul-SH3 as a novel pullulanase from Exiguobacterium sp. SH3 was cloned and expressed in both E. coli and B. subtilis. Pul-SH3 was characterized as a type I pullulanase by the conserved sequences in the primary structure as well as its substrate specificity (Li et al. 2012; Bertoldo et al. 2004). The bioinformatic analysis revealed that the enzyme shared low amino acid similarity with already known type I pullulanases. Besides, the enzyme contained two tandem CBM41 modules as a distinguishing structural feature from other type I pullulanases. The expression of the encoding gene in B. subtilis was accompanied by the secretion of the recombinant enzyme into the culture medium. Therefore, it may be assumed that signal peptides of Exiguobacterium sp. SH3 are likely to be recognized by B. subtilis secretion systems.
With an optimum activity temperature of about 45 °C and low thermal stability, Pul-SH3 may be well described as a cold-adapted pullulanase (Qoura et al. 2014). The cold-adapted enzymes are known to have higher catalytic activity at low temperatures than the mesophile and thermophile counterparts (Feller 2003). They have received an increasing attention in recent years, since their application in industry is deemed to significantly reduce costs. It has been estimated that cold hydrolysis of starch can reduce the capital and operational costs by 41 and 51 %, respectively (Cinelli et al. 2015).
Furthermore, attempts have been made to improve the thermal stability of enzymes via immobilization. Ashjari et al. (2015) reported that immobilization on silica has resulted in enhanced thermal stability of a lipase from Rhizopus oryzae. In this study, we used silica particles for immobilization of Pul-SH3 and showed that this approach can be useful for improving the thermal stability of cold-adapted enzymes. Our work is the first demonstration of thermostabilization of a cold-adapted pullulanase by immobilization on silica.
As another interesting feature, Pul-SH3 proved to be highly active over a wide range of acidic and alkaline pH. The only alkaline type I pullulanase that has been reported is S-1 pullulanase (Kim et al. 1993). According to our results, Pul-SH3 can be described as an extremely alkali-tolerant cold-adapted type I pullulanase with the ability to remain active over a broad pH range (4–11).
The tolerance against detergents is a significant characteristic by which enzymes are routinely evaluated for their potential industrial application. In this regard, the activity of pulA from Thermotoga neapolitana has been reported to drop to 17 % with 35 mM SDS (Kang et al. 2011). Also, the type I pullulanase from Fervidobacterium pennavorans Ven5, Geobacillus thermoleovorans US105, and Shewanella arctica have been reported to be totally inactivated by 1, 3.5, and 35 mM SDS, respectively (Bertoldo et al. 1999; Ayadi et al. 2008; Elleuche et al. 2015). However, Pul-SH3 in this study proved to be exceptionally tolerant against SDS and its activity was not affected by 35 mM concentration of this detergent. Even at 350 mM concentration of SDS, Pul-SH3 was able to keep about 54 % of its activity. In addition, Pul-SH3 was highly stable in the presence of 10 % Triton X-100 and Tween 20 as non-ionic detergents. It has previously been reported that the pullulanase from G. thermoleovorans completely lost its activity in the presence of 1 % of the detergents (Ayadi et al. 2008), while those from F. pennavorans and S. arctica were stable against 1 % and 10 mM Triton X-100, respectively (Bertoldo et al. 1999; Elleuche et al. 2015). According to the results of the current study, the activity of Pul-SH3 is significantly stimulated by β–mercaptoethanol (1 %) and DTT (0.2 %). Similar results have been obtained for the pullulanases from S. arctica, F. pennavorans, and T. neapolitana. The activity of Pul-SH3 was not affected by EDTA, as has been reported for F. pennavorans pullulanase (Bertoldo et al. 1999). However, it has been shown that the activity of other type I pullulanases may be either stimulated (T. neapolitana) or strongly inhibited (Bacillus cereus Nws-bc5, S. arctica) as a result of EDTA treatment. It is noteworthy that EDTA as a chelating agent inhibits the activity of cation-dependent enzymes.
In this study, the thermal stability of Pul-SH3 as a cold-adapted pullulanase was successfully improved by simple immobilization on silica support. The results obtained by this study established that Pul-SH3 is a novel cold-adapted type I pullulanase with several extremozyme characteristics, including a wide pH range of activity and high tolerance against detergents and alkali conditions. Considering the interesting characteristics of Pul-SH3, it may be assumed that the enzyme is a potential candidate for cold hydrolysis of starch.
References
Ashjari M, Mohammadi M, Badri R (2015) Chemical amination of Rhizopus oryzae lipase for multipoint covalent immobilization on epoxy-functionalized supports: modulation of stability and selectivity. J Mol Catal B Enzym 115:128–134
Ayadi DZ, Ali MB, Jemli S, Mabrouk SB, Mezghani M, Messaoud EB, Bejar S (2008) Heterologous expression, secretion and characterization of the Geobacillus thermoleovorans US105 type I pullulanase. Appl Microbiol Biotechnol 78:473–481
Bertoldo C, Duffner F, Jorgensen PL, Antranikian G (1999) Pullulanase type I from Fervidobacterium pennavorans Ven5: cloning, sequencing, and expression of the gene and biochemical characterization of the recombinant enzyme. Appl Environ Microbiol 65:2084–2091
Bertoldo C, Armbrecht M, Becker F, Schäfer T, Antranikian G, Liebl W (2004) Cloning, sequencing, and characterization of a heat- and alkali-stable type I pullulanase from Anaerobranca gottschalkii. Appl Environ Microb 70:3407–3416
Bradford M (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Chung HJ, Lee EJ, Lim ST (2002) Comparison in glass transition and enthalpy relaxation between native and gelatinized rice starches. Carbohydr Polym 48:287–298
Cinelli BA, Castilho LR, Freire DMG, Castro AM (2015) A brief review on the emerging technology of ethanol production by cold hydrolysis of raw starch. Fuel 150:721–729
Clark JH, Budarin V, Deswarte FEI, Hardy JJE, Kerton FM, Hunt AJ, Luque R, Macquarrie DJ, Milkowski K, Rodriguez A, Samuel O, Tavener SJ, White RJ, Wilson AJ (2006) Green chemistry and the biorefinery: a partnership for a sustainable future. Green Chem 8:853–860
Corre DL, Bras J, Dufresne A (2010) Starch nanoparticles: a review. Biomacromolecules 11:1139–1153
Elleuche S, Qoura FM, Lorenz U, Rehn T, Brück T, Antranikian G (2015) Cloning, expression and characterization of the recombinant cold-active type-I pullulanase from Shewanella arctica. J Mol Catal B Enzym 116:70–77
Emampour M, Noghabi KA, Zahiri HS (2015) Molecular cloning and biochemical characterization of a novel cold-adapted alpha-amylase with multiple extremozyme characteristics. J Mol Catal B Enzym 111:79–86
Feller G (2003) Molecular adaptations to cold in psychrophilic enzymes. Cell Mol Life Sci 60:648–662
Garstka MA, Fish A, Celie PHN, Joosten RP, Janssen GMC, Berlin I, Hoppes R, Stadnik M, Janssen L, Ovaa H, Veelen PAV, Perrakis A, Neefjes J (2015) The first step of peptide selection in antigen presentation by MHC class I molecules. PNAS 112:1505–1510
Hii SL, Tan JS, Ling TC, Ariff AB (2012) Pullulanase: role in starch hydrolysis and potential industrial applications. Enzyme Res 2012:1–14
Hong Y, Liu G, Gu Z (2015) Preparation and characterization of hydrophilic debranched starch modified by pullulanase on swollen granule starch. Food Res Int 67:212–218
Kang J, Park KM, Choi KH, Park CS, Kim GE, Kim D, Cha J (2011) Molecular cloning and biochemical characterization of a heat-stable type I pullulanase from Thermotoga neapolitana. Enzyme Microb Technol 48:260–266
Kar S, Ray RC, Mohapatra UB (2012) Purification, characterization and application of thermostable amylopullulanase from Streptomyces erumpens MTCC 7317 under submerged fermentation. Ann Microbiol 62:931–937
Kennedy JF, Cabalda VM, White CA (1988) Enzymic starch utilization and genetic engineering. Trends Biotechnol 6:184–189
Kim CH, Choi HI, Lee DS (1993) Pullulanases of alkaline and broad pH range from a newly isolated alkalophilic Bacillus sp. S-1 and a Micrococcus sp. Y-1. J Ind Microbiol 12:48–57
Li Y, Zhang L, Niu D, Wang Z, Shi G (2012) Cloning, expression, characterization, and biocatalytic investigation of a novel bacilli thermostable type I pullulanase from Bacillus sp. CICIM 263. J Agric Food Chem 60:11164–11172
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Bioanal Chem 31:426–428
Mojallali L, Zahiri HS, Rajaei S, Noghabi KA, Haghbeen K (2014) A novel ~34-kDa α-amylase from psychrotroph Exiguobacterium sp. SH3: Production, purification, and characterization. Biotechnol Appl Biochem 61:118–125
Nguyen QD, Rezessy-Szabó JM, Claeyssens M, Stals I, Hoschke Á (2002) Purification and characterisation of amylolytic enzymes from thermophilic fungus Thermomyces lanuginosus strain ATCC 34626. Enzyme Microb Technol 31:345–352
Plant AR, Clemens RM, Daniel RM, Morgan HW (1987) Purification and properties of a thermoactive and thermostable pullulanase from Thermococcus hydrothermalis, a hyperthermophilic archaeon isolated from a deep-sea hydrothermal vent. Appl Microbiol Biotechnol 26:427–433
Qiao Y, Peng Q, Yan J, Wang H, Ding H, Shi B (2015) Gene cloning and enzymatic characterization of alkalitolerant type I pullulanase from Exiguobacterium acetylicum. Lett Appl Microbiol 60:52–59
Qoura F, Elleuche S, Brueck T, Antranikian G (2014) Purification and characterization of a cold-adapted pullulanase from a psychrophilic bacterial isolate. Extremophiles 18:1095–1102
Rajaei S, Heidari R, Zahiri HS, Sharifzadeh S, Torktaz I, Noghabi KA (2014) A novel cold-adapted pullulanase from Exiguobacterium sp. SH3: Production optimization, purification, and characterization. Starch 66:225–234
Roy I, Gupta MN (2004) Hydrolysis of starch by a mixture of glucoamylase and pullulanase entrapped individually in calcium alginate beads. Enzyme Microb Tech 34:26–32
Saha BC, Mathupala SP, Zeikus JG (1988) Purification and characterization of a highly thermostable novel pullulanase from Clostridium thermohydrosulfuricum. Biochem J 252:343–348
Sapińska E, Balcerek M, Pielech-Przybylska K, Stanisz M (2014) The impact of treatment of cereal mashes with hydrolases of non-starch polysaccharides and pullulanase on the chemical composition of the obtained distillates. J Inst Brew 120:105–110
Wu H, Yu X, Chen L, Wu G (2014) Cloning, overexpression and characterization of a thermostable pullulanase from Thermus thermophilus HB27. Protein Expres Purif 95:22–27
Acknowledgments
This work was supported by the Iran National Science Foundation (Grant No. 91004311).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Huang.
Rights and permissions
About this article

Cite this article
Rajaei, S., Noghabi, K.A., Sadeghizadeh, M. et al. Characterization of a pH and detergent-tolerant, cold-adapted type I pullulanase from Exiguobacterium sp. SH3. Extremophiles 19, 1145–1155 (2015). https://doi.org/10.1007/s00792-015-0786-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-015-0786-6