
Abstract
Recent metagenomic analyses of Yellowstone National Park (YNP) thermal spring communities suggested the presence of minor archaeal populations that simultaneous PCR-based assays using traditional ‘universal’ 16S rRNA gene primers failed to detect. Here we use metagenomics to identify PCR primers effective at detecting elusive members of the Archaea, assess their efficacy, and describe the diverse and novel archaeal community from a circum-neutral thermal spring from the Bechler region of YNP. We determined that a less commonly used PCR primer, Arch349F, captured more diversity in this spring than the widely used A21F primer. A search of the PCR primers against the RDP 16S rRNA gene database indicated that Arch349F also captured the largest percentage of Archaea, including 41 % more than A21F. Pyrosequencing using the Arch349F primer recovered all of the phylotypes present in the clone-based portion of the study and the metagenome of this spring in addition to several other populations of Archaea, some of which are phylogenetically novel. In contrast to the lack of amplification with traditional 16S rRNA gene primers, our comprehensive analyses suggested a diverse archaeal community in the Bechler spring, with implications for recently discovered groups such as the Geoarchaeota and other undescribed archaeal groups.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydrothermal vent ecosystems have been a rich resource for the study of microbial diversity, astrobiology, and geomicrobiology for several decades. Much of the diversity in these systems is only known from environmental genomics, due to the recalcitrance of most thermal spring organisms to culturing. In particular, the study of archaeal biodiversity in hydrothermal systems has been driven by environmental genotyping, and now environmental genomics. Recent evidence, however, suggests there remains a methodological gap between the ability to detect Archaea and our understanding of their ubiquity in nature. Although universal 16S rRNA gene primers such as 21F/1492R (DeLong 1992) have lead to the discovery of many new lineages (Barns et al. 1994; DeLong 1992; Takai and Sako 1999) and transformed our view of archaeal phylogenetic diversity during the last 20 years (Barns et al. 1994; Hugenholtz 2002; Schleper et al. 2005), metagenomic studies highlight the fact that these primers are not ‘universal’. In a recent metagenomic survey of twenty Yellowstone National Park thermal springs (Inskeep et al. 2013), 16S rRNA gene clone-based surveys using 21F/1492R failed to detect Archaea in seven of the samples (Table S1). The seven springs lacking Archaea by PCR panel were all of circum-neutral to alkaline pH (6.2–9.1) and all contain minor (<1.0 % of metagenomic reads), and in some cases, greater populations of Archaea based on classification of metagenomic reads (For example, springs from the Calcite Springs and Bechler regions; Inskeep et al. 2010; Takacs-Vesbach et al. 2013; Table S1). The disparity in apparent community composition between PCR-based studies using ‘universal’ 16S rRNA gene primers and a less taxonomically biased approach, such as metagenomic analysis, suggests an inability of traditional primers to accurately represent archaeal community composition. Inaccuracy in community composition analyses, even of minor populations, can hamper efforts to discover novel biodiversity critical for evolutionary studies as well as understanding distributions of known taxa. For example, current efforts to genomically characterize low population organisms that may be evolutionary important from single cell genomics rely on accurate and complete community censuses to find genomic targets (Rinke et al. 2013).
Much initial archaeal biodiversity survey work was focused on placing novel lineages into robust phylogenetic context (Barns et al. 1994; Takai and Sako 1999), which necessitated the largest gene fragment possible, such as produced by the 21F/1492R primer combination (Olsen et al. 1986). However, with the advent of next-generation sequencing technologies, focus has shifted to high-throughput, accurate estimations of taxonomic diversity which do not require full-length gene fragments (Liu et al. 2007) and allows researchers to describe low abundance populations that may be ecologically relevant or important for evolutionary and bioprospecting studies. Many effective archaeal primers have been reported previously (for review see: Baker et al. 2003; Klindworth et al. 2013), but a robust analysis of their efficiency has not been evaluated in samples where there exists communities that were not detected by the more commonly used 21F/1492R primer combination. Further, taxonomic biases of PCR primers in the amplification of prokaryotic 16S rRNA genes in mixed communities have been previously documented in many systems (For example: Baker et al. 2003; Klindworth et al. 2013; Kumar et al. 2011; Suzuki and Giovannoni 1996). However, there has been little empirical testing of archaeal-specific PCR primers against a relatively unbiased reference, such as a metagenomic, shotgun-sequence produced dataset for a given sample. Here we tested universal archaeal 16S rRNA gene PCR primer combinations by comparing the archaeal communities described metagenomically here and in Takacs-Vesbach et al. (2013) to population results derived from complementary in silico, clone library and 454 pyrosequencing approaches.
Materials and methods
The Bechler region spring is a high temperature (~80 °C), alkaline spring (pH 7.8) that contains dense Thermocrinis spp. filament streamers along runoff channels. Archaea are also associated with the Thermocrinis filaments (Takacs-Vesbach et al. 2013). Protein-coding genes belonging to Archaea in the publically available spring metagenome (based on ≥60 % BLAST identity classifications) were analyzed using the IMG/M web interface (IMG metagenome taxon ID: 2013515002; Markowitz et al. 2012) to examine community diversity. The taxonomic affiliations of 16S rRNA genes present in the metagenomic data were assessed by phylogenetic analysis of partial 16S rRNA genes that were present for two phylotypes. 16S rRNA gene references were chosen by top uncultured clone BLAST hits in Genbank and supplemented with references for the major phylogenetic lineages of Crenarchaeota. 16S rRNA genes were aligned using pyNAST (Caporaso et al. 2010a) and trimmed so that all of the sequences shared the same homologous 691 positions common to the two metagenomic 16S rRNA gene fragments. Phylogenetic structure was explored using the MEGA software package (Tamura et al. 2013), and a final maximum likelihood tree was constructed using the GTR + G + I evolutionary model as suggested by jModelTest 2 (Darriba et al. 2012). The geochemical context of the Bechler spring and detailed microbial metagenomic analyses are described in Takacs-Vesbach et al. (2013).
Archaeal 16S rRNA genes were amplified from the Bechler spring using 11 previously published archaeal-specific primers (Table S2) with the environmental DNA used in the metagenomic analysis and in the 21F/1492R 16S rRNA gene screens reported in Takacs-Vesbach et al. (2013). PCR reactions consisted of 10 µl 5X GoTaq buffer (Promega, Madison, WI, USA), 12.5 mM dNTP (BioLine USA Inc., Taunton, MA, USA), 20 pmol of both primers, 2.5 units of GoTaq DNA polymerase (Promega), 1 µl of 2 % (w/v) bovine serum albumin, 1 mM MgCl2, and ~50 ng DNA. The PCR cycling program consisted of 30 s at 94 °C, 30 s at 55 or 58 °C (optimized for different primer pairs), and 90 s at 72 °C for a total of 30 cycles and was performed on an ABI GeneAmp 2700 (Life Technologies, Grand Island, NY, USA). Each forward primer was tested with each reverse primer, except 571F, which was tested with 1391R only, but failed to amplify any products. Successful amplifications were duplicated, combined, and gel purified with a Wizard SV gel purification kit (Promega). PCR products were cloned with the pGEM-T Easy kit (Promega). Between 13 and 18 cloned inserts, for each primer set was sequenced using the BigDye terminator cycle sequencing kit (Life Technologies) with the M13F primer on an ABI 3130x genetic analyzer (Life Technologies). Primer sets using the 1391R reverse primer produced larger bands consistent with Pyrobaculum spp. containing 16S rRNA gene introns (Itoh et al. 2003), which were gel excised and up to 8 clones were sequenced to confirm the presence/absence of Pyrobaculum spp. Clone insert identity was evaluated by aligning the cloned 16S rRNA gene sequences in mothur (Schloss et al. 2009), manually curating the alignment and measuring phylogenetic distances against reference 16S rRNA gene sequences from the metagenomic dataset and closely related Genbank clones.
Potential archaeal diversity detected by the primers was also measured by querying the RDP database of archaeal 16S rRNA genes using the Probe Match function (http://rdp.cme.msu.edu/probematch/search.jsp; Cole et al. 2014). Queries against RDP were conducted only on 16S rRNA gene sequences containing the Escherichia coli base positions encompassed by all primers (8–585 for the forward primers and 787–1407 for the reverse primers) and with 0 mismatched bases allowed. To incorporate a 16S rRNA gene dataset produced without the potential biases inherent in PCR-derived data, the primers were also tested against archaeal 16S rRNA genes present in the YNP community sequencing project described in Inskeep et al. (2013). A total of 76 archaeal 16S rRNA gene sequences from the 20 metagenomes were downloaded from the IMG/M database. The metagenome-derived 16S rRNA gene sequences were tested against each primer individually to avoid biases from comparisons to incomplete 16S rRNA gene sequences in the metagenomic contigs.
To probe the full extent of archaeal diversity in the sample, the two forward primers with the largest RDP database coverage, Arch349F and 340F, and the reverse primer A915R were used in 454 pyrosequencing analysis of the spring’s archaeal 16S rRNA gene diversity. Barcoded amplicon pyrosequencing was conducted as described previously (Andreotti et al. 2011; Dowd et al. 2008). Briefly, 100 ng of DNA per sample was amplified in triplicate by a single-step PCR to create 16S rRNA gene amplicons containing the Roche-specific sequencing adapters (454 Life Sciences, Branford, CT, USA) and a barcode unique to each sample. Amplicons were purified using Agencourt Ampure beads and combined in equimolar concentrations. Pyrosequencing was performed on a Roche 454 FLX instrument using Roche titanium reagents and titanium procedures. The 16S rRNA gene sequences were quality filtered, denoised, screened for PCR errors, and chimera checked using AmpliconNoise and Perseus to minimize potential amplification and sequencing artifacts (Quince et al. 2011). Denoised reads were classified using the Quantitative Insights Into Microbial Ecology (QIIME) package pipeline after clustering into OTUs and picking an OTU representative to assign taxonomic classification to (Caporaso et al. 2010b). OTUs were compared against references for the phylotypes found in the clone library and metagenomic datasets using a local blastn search. Raw 454 pyrosequencing data from this study are available through the NCBI Sequence Read Archive as SRP049556. The individual sff files from this study were assigned the accession numbers SAMN03145649 through SAMN03145650 under Bioproject PRJNA265119. Clone library-produced 16S rRNA gene sequence data (accessions KP091542 - KP091669) are available through Genbank.
Results and discussion
The Bechler spring metagenome contained a minor population of Archaea (8 % of the total metagenomic reads). Taxonomic classification of the assembled metagenome archaeal reads indicated a predominant Thermoproteaceae population (92 % of all archaeal reads), primarily in the genus Pyrobaculum (99 % of Thermoproteaceae reads). However, higher diversity was suggested by the classification of the remaining genes into 17 other archaeal families, including minor populations (<5 % of archaeal reads) binned in families of the phyla Euryarchaeota, Thaumarchaeota, and Korarchaeota. Only two archaeal 16S rRNA gene sequence phylotypes were present in the metagenomic dataset. One phylotype was closely related to Pyrobaculum spp. (IMG gene id YNP13_01430) with close relation to Pyrobaculum neutrophilum strain V24Sta (99 % sequence identity by BLAST, Fig. 1). The second phylotype (IMG gene ID YNP13_216600) was only closely related to a single uncultured clone from Great Boiling Spring, Great Basin, Nevada (GBS_L2_E12, Fig. 1). Both of these uncultured phylotypes were monophyletic with 16S rRNA genes from the recently described NAG1 genome (IMG taxon id 2504756013) in the proposed Geoarchaeota phylum (Kozubal et al. 2013), in addition to uncultured clones associated with Geoarchaeota from YNP thermal springs (Kozubal et al. 2012) and a phylotype closely related to the Geoarchaeota from a seafloor vent biofilm near Papua, New Guinea (clone PNG_TBR_A43; Meyer-Dombard et al. 2013). The phylum-level designation of the Geoarchaeota has been debated, as various gene combinations provide contradictory phylogenetic placement of the group within and separate from the Crenarchaeota phylum (Guy et al. 2014). Our overall archaeal topology is consistent with Kozubal et al. (2013), but differs from the sister-level relationship between Thermoproteales and Geoarchaeota using more robust phylogenetic analyses (Guy et al. 2014). Regardless, the intragroup clades were all highly supported for the Geoarchaeota. The Geoarchaeota-affiliated clones and NAG1 genome were all detected at acidic, Fe-rich YNP thermal springs (Kozubal et al. 2012, 2013), and the single seafloor vent biofilm clone was also detected in a slightly acidic, Fe-rich environment (Meyer-Dombard et al. 2013, 2012). However, both the Bechler region spring and Great Basin spring are alkaline and do not contain appreciable levels of Fe (Costa et al. 2009; Dodsworth et al. 2011; Takacs-Vesbach et al. 2013), suggesting that the phylotype detected here may constitute an ecologically and phylogenetically distinct second clade within the Geoarchaeota.

Maximum likelihood phylogenetic tree of both 16S rRNA gene sequences present in the Bechler spring metagenome. Bootstrap values are given at each node (out of 100 bootstraps) where values >50. The phylogeny is rooted with three Euryarchaeote organisms
All but one PCR primer set amplified products for the clone-based primer comparison component of the study, but only two were able to detect both archaeal 16S rRNA gene phylotypes present in the metagenomic data. A third phylotype, whose 16S rRNA gene was not recovered by the modest metagenomic sequencing effort in Takacs-Vesbach et al. (2013), was affiliated with the Aigarchaeota group of Archaea (represented by Genbank accession HM448082) and detected by many of the primer sets. All clones shared >97 % identity to one of seven phylotypes (Table 1). Both of the primer sets that amplified the three predominant phylotypes used the forward primer Arch349F. The in silico primer comparison results emphasized that the forward primers Arch21F, A109F, and A571F match sequences that, as a whole, are subsets of those matched with Arch349F and A340F, which both had the highest coverage of RDP and IMG records (Table 1; Fig. 2). This is consistent with a recent in silico analysis of archaeal primers which indicated that Arch349F had the highest overall coverage of database deposited archaeal 16S rRNA gene sequences (Klindworth et al. 2013). Reverse primers did not differ considerably in record matches (89–92 % RDP records matched by each).

Venn diagrams of 16S rRNA gene sequence record overlap detected in the RDP database by pairwise comparisons between forward primers of this study. Each primer comparison used only the 16S rRNA gene records from the RDP archaeal 16S rRNA database that covered the total range of the forward primers (E. coli base positions 8–585; 18,321 16S rRNA gene records total). Circle size and overlap is proportional to total number of records matched. Primers listed in the first column are circles on the left, and those listed in the first row are circles on the right
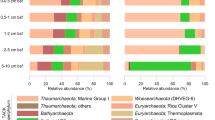
Pyrosequencing of archaeal 16S rRNA genes revealed additional novel archaeal diversity, although taxonomic differences were observed between the two primer sets that were used (Fig. 3a). A340F primarily amplified unidentified taxa within the candidate Parvarchaeota phylum (identified as the proposed order ‘Micrarchaeles’ in the latest Greengenes taxonomy), and a minor percentage of Aigarchaeota phylotypes. The Parvarchaeota are known primarily from acidic environments such as acid-mine drainage biofilms (Baker et al. 2010; Rinke et al. 2013), so it is unlikely that they are a major, active component of the Bechler community, especially considering that Crenarchaeota were the dominant phylum in the metagenomic dataset. Further, neither the Pyrobaculum sp., nor Geoarchaeota-like phylotypes that were present in the metagenomic dataset were detected by A340F, which suggests potential amplification biases. In contrast, 454 analyses with Arch349F detected all three phylotypes found in the metagenomic and clone library data in addition to several other populations. The Arch349F results are generally consistent with the metagenomic and clone library data, although the Caldiarchaceae (proposed family of the Aigarchaeota in the latest Greengenes taxonomy) populations were overrepresented relative to Pyrobaculum populations. Arch349F also detected a larger relative percentage of organisms that were not classifiable at the phylum level in addition to minor populations of unclassified Thermoprotei, Nitrosocaldus sp., Desulfurococcaceae-affiliated organisms, and Caldiarchaeum spp. not detected by A340F. Both primer sets amplified a small percentage of bacterial reads primarily classified as the family Aquificaceae, which contains the predominant microbial organism present in this community, Thermocrinis sp.(Takacs-Vesbach et al. 2013). While Arch349F only matches 1.3 × 10−5 % of RDP bacterial records (35 total of mostly unclassified bacteria), stringency could be increased with minimal loss of matched archaeal sequences by substituting a G for the K and A for the W in the 10th and 17th residues, respectively, (0/80 % and 2.7 × 10−6/94 % bacterial/archaeal records matched in RDP with 0 and 1 mismatched bases, respectively).

Bar chart showing the abundance and taxonomic classification of 454 Pyrosequencing reads for primer sets Arch349F-915R (read n = 1190) and A340F-915R (read n = 6369). Taxonomic groups are arranged in descending order by abundance in the Arch349F-915R dataset. Best classification level is given, with taxonomic hierarchy level preceding taxa name: k-kingdom, p-phyla, c-class, f-family, g-genus, and s-species. aTaxonomic classification of a shared OTU between Arch349F and the metagenomic dataset. bTaxonomic classification of a shared OTU between the clone library reference dataset and Arch349F. cTaxonomic classification of a shared OTU between the clone library reference dataset and both A340F and Arch349F
Our results suggest that commonly used primers may not be adequate for describing the archaeal diversity in thermal spring systems, and primers such as Arch349F would more accurately reflect the in situ archaeal community. Pyrosequencing with Arch349F indicated the archaeal community in this spring is taxonomically diverse, which contrasts with previous indications by unsuccessful PCR amplifications using traditional 16S rRNA gene primers and metagenomic analyses that only describe predominant taxonomic populations. While all phylotypes that were recovered by clone library and metagenomic analyses were also recovered using the forward primer Arch349F, several populations that could not be classified at or below phylum-level taxonomy were also recovered. Further, the nearly complete matching of the RDP database by Arch349F suggests that our results would likely be effective in other ecosystems. Previous database analyses of archaeal primers are concordant with our results that the Arch349F primer provides the best overall coverage of archaeal diversity and is appropriate for other short-read sequencing platforms such as illumina and Ion Torrent (Klindworth et al. 2013). In conclusion, our results suggest that leveraging next-generation sequencing and PCR primers with broader archaeal specificity provide not only greater accuracy in community composition analyses, but may aid in the detection of novel, minor populations of Archaea from environmental samples.
References
Andreotti R, Pérez de León AA, Dowd SE, Guerrero FD, Bendele KG, Scoles GA (2011) Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol 11:6
Baker GC, Smith JJ, Cowan DA (2003) Review and re-analysis of domain-specific 16S primers. J Microbiol Meth 55:541–555
Baker BJ, Comolli LR, Dick GJ, Hauser LJ, Hyatt D, Dill BD, Land ML, VerBerkmoes NC, Hettich RL, Banfield JF (2010) Enigmatic, ultrasmall, uncultivated Archaea. Proc Natl Acad Sci U S A 107:8806–8811
Barns SM, Fundyga RE, Jeffries MW, Pace NR (1994) Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc Natl Acad Sci U S A 91:1609–1613
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2010a) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010b) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM (2014) Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res 42:D633–D642
Costa KC, Navarro JB, Shock EL, Zhang CLL, Soukup D, Hedlund BP (2009) Microbiology and geochemistry of great boiling and mud hot springs in the United States Great Basin. Extremophiles 13:447–459
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
DeLong EF (1992) Archaea in coastal marine environments. Proc Natl Acad Sci U S A 89:5685–5689
Dodsworth JA, Hungate BA, Hedlund BP (2011) Ammonia oxidation, denitrification and dissimilatory nitrate reduction to ammonium in two US Great Basin hot springs with abundant ammonia-oxidizing archaea. Environ Microbiol 13:2371–2386
Dowd SE, Wolcott RD, Sun Y, McKeehan T, Smith E, Rhoads D (2008) Polymicrobial nature of chronic diabetic foot ulcer biofilm infections determined using bacterial tag encoded FLX amplicon pyrosequencing (bTEFAP). PLoS ONE 3:e3326
Guy L, Spang A, Saw JH, Ettema TJ (2014) ‘Geoarchaeote NAG1’ is a deeply rooting lineage of the archaeal order Thermoproteales rather than a new phylum. ISME J 8:1353–1357
Hugenholtz P (2002) Exploring prokaryotic diversity in the genomic era. Genome Biol 3: Reviews 0003
Inskeep WP, Jay ZJ, Tringe SG, Herrgård MJ, Rusch DB, YNP Metagenome Project Steering Committee, Working Group Members (2013) The YNP Metagenome Project: Environmental Parameters Responsible for Microbial Distribution in the Yellowstone Geothermal Ecosystem. Front Microbiol 4:67
Inskeep WP, Rusch DB, Jay ZJ, Herrgard MJ, Kozubal MA, Richardson TH, Macur RE, Hamamura N, Jennings R, Fouke BW, Reysenbach AL, Roberto F, Young M, Schwartz A, Boyd ES, Badger JH, Mathur EJ, Ortmann AC, Bateson M, Geesey G, Frazier M (2010) Metagenomes from high-temperature chemotrophic systems reveal geochemical controls on microbial community structure and function. PLoS ONE 5:e9773
Itoh T, Nomura N, Sako Y (2003) Distribution of 16S rRNA introns among the family Thermoproteaceae and their evolutionary implications. Extremophiles 7:229–233
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41:e1
Kozubal MA, Macur RE, Jay ZJ, Beam JP, Malfatti SA, Tringe SG, Kocar BD, Borch T, Inskeep WP (2012) Microbial iron cycling in acidic geothermal springs of Yellowstone National Park: integrating molecular surveys, geochemical processes, and isolation of novel Fe-active microorganisms. Front Microbiol 3:109
Kozubal MA, Romine M, Jennings R, Jay ZJ, Tringe SG, Rusch DB, Beam JP, McCue LA, Inskeep WP (2013) Geoarchaeota: a new candidate phylum in the Archaea from high-temperature acidic iron mats in Yellowstone National Park. ISME J 7:622–634
Kumar PS, Brooker MR, Dowd SE, Camerlengo T (2011) Target region selection is a critical determinant of community fingerprints generated by 16S Pyrosequencing. Plos One 6
Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R (2007) Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35:e120
Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Grechkin Y, Ratner A, Jacob B, Pati A, Huntemann M, Liolios K, Pagani I, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC (2012) IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res 40:D123–D129
Meyer-Dombard DR, Price RE, Pichler T, Amend JP (2012) Prokaryotic populations in Arsenic-Rich Shallow-Sea Hydrothermal Sediments of Ambitle Island, Papua New Guinea. Geomicrobiology J 29:1–17
Meyer-Dombard DR, Amend JP, Osburn MR (2013) Microbial diversity and potential for arsenic and iron biogeochemical cycling at an arsenic rich, shallow-sea hydrothermal vent (Tutum Bay, Papua New Guinea). Chem Geol 348:37–47
Olsen GJ, Lane DJ, Giovannoni SJ, Pace NR, Stahl DA (1986) Microbial ecology and evolution: a ribosomal RNA approach. Annu Rev Microbiol 40:337–365
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ (2011) Removing noise from pyrosequenced amplicons. Bmc Bioinform 12
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437
Schleper C, Jurgens G, Jonuscheit M (2005) Genomic studies of uncultivated archaea. Nat Rev Microbiol 3:479–488
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing mothur: Open-source, Platform-Independent, Community-supported Software for Describing and Comparing Microbial Communities. Appl Environ Microbiol 75:7537–7541
Suzuki MT, Giovannoni SJ (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630
Takacs-Vesbach C, Inskeep WP, Jay ZJ, Herrgard MJ, Rusch DB, Tringe SG, Kozubal MA, Hamamura N, Macur RE, Fouke BW, Reysenbach A-L, McDermott TR, Jennings Rd, Hengartner NW, Xie G (2013) Metagenome sequence analysis of filamentous microbial communities obtained from geochemically distinct geothermal channels reveals specialization of three aquificales lineages. Front Microbiol 4
Takai K, Sako Y (1999) A molecular view of archaeal diversity in marine and terrestrial hot water environments. FEMS Microbiol Ecol 28:177–188
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729
Acknowledgments
This work was funded by National Science Foundation (NSF) DEB0206773 and the Department of Energy-Joint Genome Institute Community Sequencing Program (CSP 787081). DC received additional support from NSF EHR0832947, RT from National Institutes of Health (NIH) R25 GM075149, and KM from the New Mexico Space Grant Consortium. Technical support for sequencing was provided by the University of New Mexico’s Molecular Biology Facility, which is supported by NIH grant P20GM103452 from the Institute Development Award Program of the National Center for Research Resources. We thank W. Inskeep for comments on the manuscript and providing PCR panel success results used in Table S1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Oren.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Colman, D.R., Thomas, R., Maas, K.R. et al. Detection and analysis of elusive members of a novel and diverse archaeal community within a thermal spring streamer consortium. Extremophiles 19, 307–313 (2015). https://doi.org/10.1007/s00792-014-0715-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-014-0715-0