
Abstract
A novel alkaliphilic esterase (EstJ) was identified from a soil metagenome of Jeju Island, Korea, using a 96-well plate-based functional assay for determination of pH dependence of activity. The amino acid sequence of EstJ showed low similarity (32–45 %) to putative α/β hydrolases derived from whole-genome sequencing studies. EstJ, although not belonging to any of the known families of bacterial lipolytic enzymes, however, it showed closest sequence identity to the family IV enzymes that are related to the mammalian hormone-sensitive lipases. The highly conserved motifs of family IV enzymes were found in EstJ, but the corresponding sequences of each motif in EstJ were unique; most particularly the –(F/Y)(F/Y/L)HGGG– motif was represented by –WMVSGG–. The purified EstJ was highly active from pH 8.5 to 10.5. More than 90 % of maximum activity was also retained over a wide pH range of 5.5–0.5 after prolonged incubation. EstJ was also moderately thermophilic with an optimum temperature of 55 °C. Therefore, EstJ is the first metagenome-derived bacterial family IV esterase possessing both highly alkaliphilic and moderately thermophilic properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lipolytic enzymes such as lipase (triacylglycerol acylhydrolases, EC 3.3.3.3) and esterase (carboxylic ester hydrolases, EC 3.1.1.1) are some of the most versatile and important biocatalysts in industrial biotechnology applications (Jaeger and Eggert 2002; Panda and Gowrishankar 2005). Because of their ability to catalyze numerous bioconversion reactions such as hydrolysis, alcoholysis, acidolysis, esterification, transesterification, interesterification and aminolysis, these enzymes have been used for the production of oleochemicals, cosmetics, pharmaceuticals and other organic chemicals (Houde et al. 2004; Kapoor and Gupta 2012). Therefore, bacterial esterases and lipases with different characteristics are constantly sought. Among them, a number of family IV lipolytic enzymes that display a striking sequence and structural similarity to mammalian hormone-sensitive lipases have been identified from bacteria and metagenome libraries (Hårdeman and Sjöling 2007; Ko et al. 2012; Rhee et al. 2005; Roh and Villatte 2008). These family IV enzymes have diverse biochemical characteristics, several of which are psychrophilic (Hårdeman and Sjöling 2007; Ko et al. 2012; Roh and Villatte 2008) or thermophilic (Rhee et al. 2005).
A metagenome, the total genetic material recovered from environmental samples, includes the genomes from as-yet uncultured or uncultivable microorganisms. By enabling us to harness the natural genetic diversity present in the environment (Simon and Daniel 2009), a metagenomic DNA library plays an important role in the identification of new enzymes with unique properties (Schmeisser et al. 2007; Steele et al. 2009). To date, many esterases and lipases have been identified from metagenomic DNA libraries and biochemically characterized. Among them, only a small number of esterases and lipases have been found to be alkaliphilic; an alkaliphilic enzyme is one that functions optimally in alkaline pH values. The metagenome-derived alkaliphilic enzymes include Est2K from a compost metagenome (Kim et al. 2010), LipEH166 from an intertidal flat sediment metagenome (Kim et al. 2009), EM2L8 esterase from a deep-sea sediment metagenome (Park et al. 2007), EstCE1 from an oil-contaminated soil metagenome (Elend et al. 2006), EstA3 from a drinking water biofilm metagenome (Elend et al. 2006), and CHA2 from an Antarctic soil metagenome (Hu et al. 2012).
In this study, we describe the isolation, sequence analysis and enzymatic characterization of a novel alkaliphilic esterase EstJ from a soil metagenomic DNA library. According to the sequence analysis, the alkaliphilic EstJ was similar to bacterial family IV enzymes, but has a distinctive feature in its essential sequence motifs. Moreover, EstJ possesses an alkaliphilic and moderately thermophilic mode of activity and showed very high stability particularly over a broad pH range.
Materials and methods
Bacterial strains, plasmids, and environmental samples
Environmental soil samples with different local geological features were collected from five different places on the volcanic Jeju Island, South Korea, including soils from a parasitic volcano wetland (Mulyeongari-Oreum; 33°37′N, 126°69′E), seashore sediments (Hanlim Harbor; 33°41′N, 126°26′E), coastal rocks (Oedolgae Rock; 33°24′N, 126°54′E) and a waterfall (Cheonjiyeon Falls; 33°25′N, 126°55′E), and were used to construct a metagenomic DNA library in Escherichia coli EPI100-T1R, a T1 phage-resistant strain (Epicentre Biotechnologies). The fosmid EpiFOS™-5 (Epicentre) was used as a vector for constructing the metagenomic DNA libraries. E. coli strain XL1-Blue (RBC Bioscience) and plasmids pUC19 and pCR2.1-TOPO (Invitrogen) were used for the cloning of the metagenomic esterase-encoding gene. E. coli strain BL21(DE3) (RBC Bioscience) and plasmid pET21a (Novagen) were used for the recombinant expression of the esterase.
Construction of metagenomic library from soil samples
The DNA extraction solution used to isolate the metagenomic DNA from the soil samples contained 100 mM Tris–HCl (pH 8.0), 10 mM ethylenediaminetetraacetic (EDTA), 10 mM sodium phosphate (pH 8.0), 1.5 M NaCl, 1 % (w/v) hexadecyl trimethyl ammonium bromide, and 50 μg/μl of proteinase K. After 13.5 ml of DNA extraction solution was added to 5 g of soil samples, the tube was incubated at 37 °C for 30 min with gentle shaking. Sodium dodecyl sulfate (SDS) was added to a final concentration of 2 % and then, the mixture was further incubated at 65 °C for 2 h. After centrifugation at room temperature, the supernatant was collected and an equal volume of chloroform/isoamyl alcohol (24:1) was added to the supernatant. Protein and cell debris were removed from the solution by centrifugation. After the DNA extraction procedure was repeated three times, 0.6 volume of isopropanol was added to the clear lysate sample and the mixture was incubated for 60 min at room temperature. The metagenomic DNA was precipitated by centrifugation at 12,000×g for 20 min at 20 °C, washed in 70 % ethanol and resuspended in an appropriate volume of distilled water. Finally, the metagenomic DNA was purified by preparative low melting agarose gel electrophoresis, and end-repaired (DNA End-Repair kit, Epicentre). The next steps to construct the metagenomic DNA library in E. coli were carried out using a EpiFOS™-5 vector and Lambda packaging solution, according to manufacturer’s protocols (pEpiFOS™-5 fosmid library production kit, Epicentre). Pulse-field gel electrophoresis with a CHEF-DRIII system (Bio-Rad Laboratories, Richmond, CA, USA) was used to analyze the purified metagenomic DNA and the restriction digest of fosmid–insert DNA.
Screening of metagenomic DNA library for alkaliphilic esterase activity
An appropriate volume of aliquot from the frozen stock of the metagenomic library was transferred to each Luria–Bertani (LB) agar plate containing 0.8 % (w/v) tributyrin and 34 μg/ml chloramphenicol (Cm). The plates were incubated at 37 °C for 20 h, and the colonies surrounded by a transparent halo zone were transferred to 96-well plates containing 800 μl of LB/Cm broth. After the 16-h incubation at 37 °C, 100 μl of the culture was transferred to fresh 96-well plates containing 800 μl of LB/Cm broth and 1 μl of CopyControl Fosmid Autoinduction Solution (Epicentre). Cells were grown at 37 °C for 4 h with vigorous agitation (Heidolph Titramax 1000, Heidolph Electro, Germany), harvested by centrifugation at 4 °C, and finally disrupted by addition of BugBuster Plus Benzonase Nuclease (Novagen). After the cell debris was removed by centrifugation, the supernatant was transferred to a set of 96-well plates, and then different pH-buffered substrate solutions (10 mM p-nitrophenyl butyrate; 50 mM Tris–HCl for pH 7.0 and 50 mM Glycine-NaOH for pH 10.0) were added to each of the 96-well plates containing 50 μl of the cell extract. The metagenomic E. coli clone showing alkaliphilic esterase activity was selected based on the ratio of esterase activity at pH 10.0 relative to that at pH 7.0.
Subcloning and DNA sequencing
The fosmid DNA was isolated using the FosmidMAX DNA purification kit (Epicentre) and then digested with the restriction enzyme Sau3AI. DNA fragments of 3–5 kb were recovered using agarose gel electrophoresis and ligated into the BamHI site of the pUC19 vector. The ligated DNA was used to transform E. coli XL-Blue cells. The transformed E. coli showing lipolytic activity was selected on LB agar plates containing 0.8 % tributyrin, and the plasmid was isolated from the culture of positive E. coli cells. The DNA sequences of the metagenomic insert of the isolated plasmid were determined using the primer-walking technique by Macrogen Inc. (Daejeon, Korea). Sequence analysis, database search, and gene structure characterization were carried out using programs and databases at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). The signal sequence of the enzyme was predicted using SignalP version 4.0 (http://www.cbs.dtu.dk/services/SignalP/), and the molecular weight and pI of the enzyme were predicted using the ExPASy Compute pI/Mw tool (http://web.expasy.org/compute_pi/). A phylogenetic tree of protein sequences was generated using ClustalW with the MegAlign program (DNAstar).
Esterase expression and purification
The EstJ-encoding gene was amplified by PCR using the primers Lig1F (5′-CATATGAAGCTCTCTACAAGCAA-3′) and Lig1R (5′-CTCGAGTCATTTCTGCGCT-3′), which contained the restriction enzyme sites for NdeI and XhoI, respectively. After a standard PCR was performed, the PCR products were cloned into the pCR2.1-TOPO vector (Invitrogen) and then sequenced to ensure the correct gene amplification. The NdeI- and XhoI-digested fragments were prepared and ligated into pET-21a. The recombinant plasmid was introduced into E. coli, and the transformant was grown in LB broth containing 50 μg/ml ampicillin at 37 °C until the optical density reached to 0.6 at 600 nm. Induction was started by adding 0.5 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) with shaking at 18 °C. After 20 h, the cells were harvested by centrifugation and washed with 50 mM of Tris–HCl (pH 8.0). The cell pellet was resuspended in IMAC lysis solution consisting of 300 mM of KCl, 50 mM of KH2PO4, and 5 mM of imidazole (pH 8.0; Bio-Rad) and homogenized using an ultrasonic homogenizer (Sonics & Materials, Danbury, CT, USA). The 6X histidine-tagged enzyme was purified using a Profinia™ Protein purification system equipped with a Bio-Scale Mini Profinity IMAC cartridge and a Bio-Scale Mini Bio-Gel P-6 desalting cartridge (Bio-Rad Laboratories). The protein concentration was measured using Bradford protein assay kit (Bio-Rad Laboratories). The purity and molecular weight of the purified protein were confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Enzyme assay
The enzymatic activity was measured by a spectrophotometric method using p-nitrophenyl esters as described previously (Yu et al. 2011). In brief, the reaction started with the addition of enzyme to an appropriate amount of the following reaction mixture: 1 mM p-nitrophenyl ester, 1 % (v/v) acetonitrile, 4 % (v/v) ethanol, and 47.5 mM Tris–HCl (pH 8.0). The absorbance of the liberated p-nitrophenol was continuously monitored for 10 min at 405 nm with the Multiskan UV/Vis microplate spectrophotometer (Thermo Scientific). The esterase activity was calculated by comparing the sample values to a standard curve drawn with different concentrations of p-nitrophenol. One unit of enzyme activity was defined as the amount of enzyme required to release 1 μmol of p-nitrophenol per minute from the p-nitrophenyl ester.
Biochemical properties
The same amount of purified enzyme was used in each experiment to analyze the biochemical properties, with all experiments performed in triplicate. Substrate preference was examined by assaying the hydrolytic activities for p-nitrophenyl esters of varying acyl chain lengths from C2 to C18 at 37 °C and pH 8. To determine the kinetic parameters of enzyme catalysis, the initial reaction velocities were determined at different substrate concentrations (up to 0.4 mM), and fitted to the Lineweaver–Burk transformation of the Michaelis–Menten equation. The optimal temperature for esterase activity was determined by assaying the hydrolysis of p-nitrophenyl caproate at different temperatures (10–70 °C). To determine the enzymatic thermostability, the residual activity was measured after the enzyme was preincubated at different temperatures (10–65 °C) for various periods (0–60 min). The effect of pH on catalytic activity was examined at 37 °C using different pH buffers (pH 4.5–6.0, sodium citrate-citric acid; pH 6.0–8.0, sodium phosphate (monobasic and dibasic); pH 8.0–9.0, sodium tetraborate-HCl; pH 9.0–10.0, glycine-NaOH; pH 10.0–11.0, sodium bicarbonate-NaOH; pH 11.0–12.0, disodium hydrogen orthophosphate-NaOH). To ensure the accuracy of the pH buffers used, the pH electrode was calibrated at the temperature to be used for all the measurements in advance. Because the extinction coefficient of p-nitrophenol is altered depending on the buffer pH, a standard curve was determined at each pH value. It was also taken into consideration that non-enzymatic spontaneous hydrolysis of p-nitrophenyl esters occur at extreme pH values. To determine the enzymatic stability under different pH values, the enzyme solution was preincubated in different pH buffers (50 mM) ranging from 4.5 to 12.0 for 1 and 24 h; a nine-fold volume of buffers was mixed with one volume of enzyme solution. The enzyme solution was then mixed with the same volume of 50 mM Tris–HCl buffer (pH 8.0), and for the measurement of the residual activity, 50 μl of the solution was added to 950 μl of the assay mixture consisting of 1 mM p-nitrophenyl caproate, 1 % (v/v) acetonitrile, 4 % (v/v) ethanol, and 47.5 mM Tris–HCl (pH 8.0).
The effects of divalent ions including CaCl2, MgCl2, ZnSO4, and CuSO4 and the chelating agent EDTA on enzyme activity were examined by adding them to the assay solution at final concentrations of 1, 5, and 10 mM, respectively. The activity was then measured at 37 °C and pH 8.0. Enzymatic stability in water-miscible organic solvents such as methanol, ethanol, dimethyl sulfoxide, and acetonitrile was examined after 1-h incubation of enzyme at 37 °C and 10 % organic solvent (final). The residual activity was measured at 37 °C and pH 8.0 by adding a portion of the enzyme solution to the assay solution. For the measurement of enzyme stability in methanol, the residual activity was further monitored after the enzyme was incubated in various concentrations (up to 50 %) of methanol.
Results and discussion
Screening and isolation of alkaliphilic esterase from metagenome
In total, about 60,000 clones of E. coli containing the fosmid-based metagenomic DNA library were prepared using soil samples from five different locations on Jeju Island, South Korea. Restriction analysis of five recombinant fosmids randomly selected from each library were carried out with the restriction endonuclease BamHI, and showed that the average insert size of the fosmid clones was estimated to be about 32 kbp (data not shown).
The high-throughput two-step screening process was carried out to obtain a lipolytic enzyme with alkaliphilic properties from the metagenome. First, functional expression screening of the soil metagenomic DNA library in E. coli was done based on the halo-forming activity of the E. coli host cells containing a lipolytic enzyme-encoding gene. The first screening resulted in 121 positive E. coli colonies when they were grown on the tributyrylglycerol agar plates and further incubated at 4 °C. Second, the crude cell extract of the positive E. coli cultures was prepared and examined to determine the ratio of enzyme activity under the assay conditions at pH 10.0 to that at pH 7.0 (Fig. S1 in supplementary material). The ratio of activity at 10.0–pH 7.0 was under 1.0 in most cell extract samples, and only seven of the 121 positive fosmid clones had a ratio of more than 1, indicating alkaliphilic activity. We identified a single positive E. coli exhibiting far stronger activity at alkaline pH (an activity ratio of 3.91) than the others; the positive E. coli originated from the metagenome of the soil sample from Hanlim Harbor, Korea. After a subclone library was constructed with the fosmid conferring the alkaliphilic lipolytic activity, the plasmid (pM-15P-10sEST) containing the approximate 3.6-kb metagenomic insert was finally obtained on the tributyrylglycerol agar plates and sequenced from both ends.
Sequence analysis of EstJ
We identified an ORF of 954 bp, which we designated estJ, from the metagenomic DNA sequences of the plasmid pM-15P-10sEST (GenBank accession no. JQ807667). The esterase gene encoded a protein of 317 amino acids with a predicted molecular weight of 34 kDa and an isoelectric point of 7.6. The first 29 residues of the esterase EstJ were predicted to be a cleavable signal peptide using the prediction program SignalP on the condition that the metagenomic enzyme was derived from either a Gram-negative or Gram-positive bacteria. The mature EstJ enzyme had a predicted molecular weight of 32 kDa and a predicted pI of 7.0.
BLAST searches and amino acid sequence alignments revealed that the metagenomic EstJ was most similar to seven proteins of the α/β hydrolase superfamily from the recently annotated whole-genome sequences and one lipolytic protein from activated sludge samples (Table S1 in Supplementary material). However, EstJ shared only a low similarity (32–45 %) with the putative α/β hydrolases; none of these proteins have been experimentally examined.
A phylogenetic tree was generated for EstJ and the bacterial lipolytic enzymes representing eight different families (Arpigny and Jaeger 1999). As shown in Fig. 1, EstJ and its most closely related protein sequences did not belong to any of the known families. They were most closely related to the family IV enzymes that display a prominent amino acid sequence similarity to mammalian hormone-sensitive lipases. The family IV enzymes have four conserved sequence motifs, namely the oxyanion hole-forming residues –HGGG–, the pentapeptide –GDSAG– signature motif with a serine acting as the catalytic nucleophile, and two C-terminal conserved motifs, –DPLR– and –HGF– (Arpigny and Jaeger 1999). We then retrieved the bacterial family IV carboxylesterase sequences reported to date and performed a multiple sequence alignment. With regard to the presence of the four conserved sequence motifs and their location within the sequence, EstJ was similar to all the representative family IV enzymes (Fig. 2a). The putative catalytic triad residues, Ser150, Asp255, and His287, were also identified within these motifs of EstJ. However, EstJ and its most homologous proteins varied in their amino acid composition within each motif, particularly in that the –VSGG– sequence at position 77–80 of EstJ replaced the –HGGG– oxyanion hole-forming residues of the family IV enzymes (Fig. 2b).

Phylogenetic analysis of EstJ and closely related proteins. The phylogenetic tree was generated using the neighbor-joining method (MEGA4.0 software). The protein sequences of previously identified enzymes were retrieved with the accession numbers for the GenBank database. The numbers at the nodes indicate the bootstrap percentages of 1,000 replicates

Sequence alignment of EstJ with the conserved amino acid sequence blocks of the bacterial family IV lipolytic enzymes (a) and its four most homologous sequences (b). a The representative sequences of bacterial family IV enzymes were chosen from the search results for research articles, and then retrieved from GenBank with the following accession numbers: YP_442879, lipase/esterase from Burkholderia thailandensis E264; ZP_00943646, esterase from Ralstonia solanacearum UW551; AAC38151, lipase from Pseudomonas sp. B11-1; 1EVQ_A, carboxylesterase Est2 from Alicyclobacillus acidocaldarius; AAB89533, carboxylesterase (EstA) from Archaeoglobus fulgidus DSM 4304; AAC41424, lipase-like enzyme from Cupriavidus necator; and NP_005348, hormone-sensitive lipase from homo sapiens. b The amino sequences of homologs were identified by a BLAST search of the amino acid sequence of EstJ and the corresponding GenBank accession numbers of the sequences are as follows: alpha/beta hydrolase domain-containing protein_3 from Pedosphaera parvula Ellin514; ZP_03632535, Esterase/lipase-like protein from Spirosoma linguale DSM 74; YP_003389158, Lipase/esterase ORF12A from Uncultured sludge bacterium; ADC79146; and Alpha/beta hydrolase from Planctomyces maris DSM 8797; ZP_01853484. Identical and similar amino acid residues are shaded in black and gray, respectively. The characteristic motif and the catalytic triad residues of family IV enzymes are indicated by boxes and circles, respectively
One or both of Phe and Tyr are generally located just before the His residue of the –HGGG– motif in typical family IV enzymes. This observation also holds true for most of the metagenome-derived family IV enzymes sequenced to date (Fig. S2 in Supplementary material). Interestingly, EstJ and the homologous proteins shown in Fig. 2b had the –(V/L)SGG– motif, and in the case of EstJ, the -Trp-Met- residues were located at positions 75–76. It has been reported that the indole group of Trp exerts various functions in some proteins, for example, including an influence on substrate binding in E. coli thioesterase I (EC 3.1.2.2) (Lee et al. 2009) and an ideal positioning of the active site residues of Pseudomonas aeruginosa OXA-10 β-lactamase (EC 3.5.2.6) (Baurin et al. 2009). The Trp23 residue in the oxyanion hole of E. coli thioesterase I was suggested to play a role in interlinking the residues participating in the catalytic charge relay (Ser, Asp and His) and oxyanion hole stabilization. The Trp154 residue in the active site omega-loop of P. aeruginosa OXA-10 β-lactamase was suggested to play a role in the activity and stability of the enzyme by the interaction between the indole group of Trp154 and a conserved carboxylated Lys residue in the active site. Therefore, we suggest that the Trp75 residue of the –WMVSGG– sequence could play a significant role in the catalytic properties of EstJ, although further precise investigation such as a study on the effects of mutating the Trp residue is required.
Biochemical characterization of EstJ
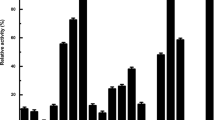
The 6X histidine-tagged EstJ was highly purified with a yield of 38 % and a purification fold of 31 (Table S2 in Supplementary material). The purified enzyme appeared as a strong single band with the densitometric purity reaching almost 95 % (Fig. 3). EstJ efficiently hydrolyzed the ester bonds of the p-nitrophenyl esters and triglycerides with acyl chains shorter than C10, but showed activity ranging from less than 5 % to none toward medium- and long-chain acyl substrates (Fig. 4). In particular, the enzyme showed the highest activity toward p-nitrophenyl butyrate (C4) with a specific activity of 4.8 U/mg under standard assay conditions of 37 °C and pH 8. When the kinetic parameters were determined for p-nitrophenyl butyrate, the initial reaction velocities of the enzyme obeyed Michaelis–Menten kinetics (Fig. S3 in Supplementary material). The K m, k cat, and k cat/K m values of EstJ were 1.06 mM, 29,600 s−1, and 27,900 s−1mM−1, respectively.

SDS-PAGE analysis of the expression and purification of EstJ. Lane M molecular weight standards, 1 total cellular extracts before induction, 2 total cellular extracts after induction, 3 insoluble fraction of cellular extracts, 4 soluble fraction of cellular extracts, 15 purified enzyme

Substrate specificity of EstJ. The activity of the purified EstJ was examined using p-nitrophenyl esters with acyl chains of different lengths. The activity was highest when employing p-nitrophenyl butyrate as the substrate, and the value was taken as 100 %. The activity values that the enzyme had at pH 9.5 (a) and 7.5 (b), respectively, were taken as 100 %
EstJ was highly active between 40 and 60 °C, retaining at least 65 % of the maximum activity at 55 °C. The enzyme activity markedly decreased at temperatures more than 60 °C (Fig. 5a). Thermostability analysis showed that EstJ remained highly stable at temperatures less than 50 °C, and could retain almost 100 % of the original activity after 60 min incubation. After incubation at 50 °C for 60 min, EstJ lost approximately 40 % of its activity, but it was extremely thermolabile at temperatures above 60 °C (Fig. 5b). The temperature-dependent activity and stability analysis indicated that EstJ had moderately thermophilic properties including a higher optimum temperature than that of a typical mesophilic enzyme. Family IV lipolytic enzymes with a high degree of thermoactivity and thermostability have been identified from thermophilic archea (Hotta et al. 2002) and the metagenome of thermal environmental samples (Rhee et al. 2005). Therefore, we proposed that EstJ with a certain degree of thermophilicity may have originated from the metagenome of a thermophilic microorganism.

Effect of temperature on the activity (a) and the stability (b) of EstJ. The values of activity that the enzyme showed at 55 °C (a) and without incubation (b), respectively, were taken as 100 %. The means and the standard deviations were calculated based on three independent experiments
To determine the pH dependence of enzymatic activity and stability, the assay was conducted under different pH-buffered conditions over the pH range of 4.5–12. The maximum activity of EstJ was observed at pH 9.5, and nearly 80 % of maximum activity was still exhibited at pH 9.0 and 10.0 (Fig. 6a). In other words, it was relatively more active at the alkaline pH values of 8.5–10.5, retaining more than 60 % of maximum activity. The highest activity was shown at pH 9.5, which was about two- and fivefold higher than the activity shown at pH 7.0 and 6.0, respectively. This observed ratio of activity at the optimum alkaline pH to the activity at other pH values is variable as is the range over which the activity is stable, depending on the enzyme. For example, the CHA2 esterase from an arctic soil metagenome had the highest activity at pH 11.0, while it decreased rapidly under pH 10.0; the activity at pH 10.0 was only about half of the activity at pH 11.0 (Hu et al. 2012). Comparatively, the pH range over which the EstCE1 esterase was highly active (Elend et al. 2006) was similar to that of EstJ.

Effect of pH on the activity (a) and the stability (b) of EstJ. The purified enzyme samples were incubated in buffers of different pH values varying from 4.5 to 12.0 for 24 h, and the residual activities were measured at pH 8.0 (b). The activity values at pH 9.5 (a) and 7.5 (b), respectively, were taken as 100 %. The means and the standard deviations were calculated based on three independent experiments. The series of different pH buffer solutions are listed in the “Materials and methods”
Analysis of pH stability also indicated that after 60 min incubation, EstJ was highly stable over the alkaline pH range up to pH 10.5. Moreover, even after a prolonged 24-h incubation of EstJ, high stability (>90 % of maximum stability) of EstJ was retained over a wide pH range of 5.5–10.5 (Fig. 6b). When considering the biochemical properties of family IV enzymes and other lipolytic enzymes, it was very interesting that a relatively high level of enzyme stability was maintained over a broad pH range. Judging from its pH optima of 9.5 and its extreme stability over a broad pH range including alkaline pH values, it was likely that the esterase EstJ-encoding gene was derived from the metagenomic DNA of an alkaliphile. Furthermore, to the best of our knowledge, the metagenomic EstJ is the first enzyme with high alkaliphilic properties in the bacterial family IV enzymes that has been characterized experimentally. For example, low-temperature adapted (Hårdeman and Sjöling 2007; Ko et al. 2012; Roh and Villatte 2008) and thermophilic family IV enzymes (Rhee et al. 2005) have been identified from a metagenome, but none of them showed such marked alkaliphilic properties as observed for EstJ. Furthermore, while many other alkaliphilic esterases have been isolated and characterized from alkaliphiles as well as from metagenomes, the stability of EstJ over the broad pH range is very unusual.
A valuable biocatalyst that works under extreme environmental conditions has typically been obtained from extremophiles such as thermophiles and hyperthermophiles, psychrophiles, halophiles, alkalophiles/acidophiles, and solvent-resistant microorganisms. In fact, from currently identified lipases and esterases with alkaliphilic properties, a number of alkalophiles were explored (Fuciños et al. 2012). However, we believe that the metagenomic approach to identifying such enzymes was not always restricted to employing the physicochemical conditions of the environmental samples where the metagenome was extracted. The alkaliphilic EstJ discussed herein originated from the metagenome of a soil sample (pH 6.8) that was not directly connected with alkaline environments. Most of the metagenome-derived alkaliphilic lipases and esterases were also derived from an ordinary environmental sample that was not identified as being alkali in nature (Elend et al. 2006; Park et al. 2007; Kim et al. 2009; Kim et al. 2010; Hu et al. 2012).
EstJ activity was severely inhibited by 1, 5 and 10 mM of divalent ions such as CaCl2 and CuSO4, but was not affected by the same concentrations of MgCl2 (data not shown). The addition of the chelating agent EDTA (1 and 5 mM) resulted in no significant reduction in the EstJ activity. Therefore, it was obvious that divalent metal ions were unnecessary for the catalytic activity of EstJ. The presence of non-ionic surfactants (0.1 and 1.0 %) such as Triton X-100 and Tween 80 increased the activity without reducing the enzyme stability. It was often observed in other lipolytic enzymes. For example, 1 % (v/v) of Tween– and Triton X–series detergents enhanced catalytic activity of esterase to 5–30 folds (Kim et al. 2006). High stability of esterase and lipase in organic solvents had an advantage in the synthesis of fatty acid alkyl esters (Röttig et al. 2010). Tolerance of EstJ to water-miscible organic solvents was examined at concentrations of 10 % (v/v). The enzyme was stable in the presence of methanol, ethanol, dimethyl sulfoxide and acetonitrile, retaining at least 90 % of its original activity. Moreover, nearly 80 % of the original activity of EstJ was still retained after 60 min incubation in up to a 30 % methanol-aqueous solution, but the enzyme activity rapidly decreased during incubation in 40 and 50 % methanol (Fig. S4 in Supplementary material).
Conclusion
Exploration of metagenomic libraries was effective in identifying a novel lipolytic enzyme with desired properties. Metagenomic libraries were screened for an alkaline-active lipolytic enzyme and the alkaliphilic esterase EstJ, which was highly stable over a wide pH range, was identified and classified as a new subgroup of bacterial lipolytic enzymes. Because there is a demand for alkaliphilic enzymes for many different applications such as environmentally safe detergent formulations, pharmaceuticals, agrochemicals, biopolymers and biodiesel, we are currently developing a genetic variant of EstJ with catalytic capabilities suitable for industrial purposes.
References
Arpigny JL, Jaeger KE (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Baurin S, Vercheval L, Bouillenne F, Falzone C, Brans A, Jacquamet L, Ferrer JL, Sauvage E, Dehareng D, Frère JM, Charlier P, Galleni M, Kerff F (2009) Critical role of tryptophan 154 for the activity and stability of class D beta-lactamases. Biochemistry 48:11252–11263
Elend C, Schmeisser C, Leggewie C, Babiak P, Carballeira JD, Steele HL, Reymond JL, Jaeger KE, Streit WR (2006) Isolation and biochemical characterization of two novel metagenome-derived esterases. Appl Environ Microbiol 72:3637–3645
Fuciños P, González R, Atanes E, Sestelo AB, Pérez-Guerra N, Pastrana L, Rúa ML (2012) Lipases and esterases from extremophiles: overview and case example of the production and purification of an esterase from Thermus thermophilus HB27. Methods Mol Biol 861:239–266
Hårdeman F, Sjöling S (2007) Metagenomic approach for the isolation of a novel low-temperature-active lipase from uncultured bacteria of marine sediment. FEMS Microbiol Ecol 59:524–534
Hotta Y, Ezaki S, Atomi H, Imanaka T (2002) Extremely stable and versatile carboxylesterase from a hyperthermophilic archaeon. Appl Environ Microbiol 68:3925–3931
Houde A, Kademi A, Leblanc D (2004) Lipases and their industrial applications: an overview. Appl Biochem Biotechnol 118:155–170
Hu XP, Heath C, Taylor MP, Tuffin M, Cowan D (2012) A novel, extremely alkaliphilic and cold-active esterase from Antarctic desert soil. Extremophiles 16:79–86
Jaeger KE, Eggert T (2002) Lipases for biotechnology. Curr Opin Biotechnol 13:390–397
Kapoor M, Gupta MN (2012) Lipase promiscuity and its biochemical applications. Process Biochem 47:555–569
Kim YJ, Choi GS, Kim SB, Yoon GS, Kim YS, Ryu YW (2006) Screening and characterization of a novel esterase from a metagenomic library. Protein Expr Purif 45:315–323
Kim EY, Oh KH, Lee MH, Kang CH, Oh TK, Yoon JH (2009) Novel cold-adapted alkaline lipase from an intertidal flat metagenome and proposal for a new family of bacterial lipases. Appl Environ Microbiol 75:257–260
Kim YH, Kwon EJ, Kim SK, Jeong YS, Kim J, Yun HD, Kim H (2010) Molecular cloning and characterization of a novel family VIII alkaline esterase from a compost metagenomic library. Biochem Biophys Res Commun 393:45–49
Ko KC, Rim SO, Han Y, Shin BS, Kim GJ, Choi JH, Song JJ (2012) Identification and characterization of a novel cold-adapted esterase from a metagenomic library of mountain soil. J Ind Microbiol Biotechnol 39:681–689
Lee LC, Chou YL, Chen HH, Lee YL, Shaw JF (2009) Functional role of a non-active site residue Trp(23) on the enzyme activity of Escherichia coli thioesterase I/protease I/lysophospholipase L(1). Biochim Biophys Acta 1794:1467–1473
Panda T, Gowrishankar BS (2005) Production and applications of esterases. Appl Microbiol Biotechnol 67:160–169
Park HJ, Jeon JH, Kang SG, Lee JH, Lee SA, Kim HK (2007) Functional expression and refolding of new alkaline esterase, EM2L8 from deep-sea sediment metagenome. Protein Expr Purif 52:340–347
Rhee JK, Ahn DG, Kim YG, Oh JW (2005) New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl Environ Microbiol 71:817–825
Roh C, Villatte F (2008) Isolation of a low-temperature adapted lipolytic enzyme from uncultivated micro-organism. J Appl Microbiol 105:116–123
Röttig A, Wenning L, Bröker D, Steinbüchel A (2010) Fatty acid alkyl esters: perspectives for production of alternative biofuels. Appl Microbiol Biotechnol 85:1713–1733
Schmeisser C, Steele H, Streit WR (2007) Metagenomics, biotechnology with non-culturable microbes. Appl Microbiol Biotechnol 75:955–962
Simon C, Daniel R (2009) Achievements and new knowledge unraveled by metagenomic approaches. Appl Microbiol Biotechnol 85:265–276
Steele HL, Jaeger KE, Daniel R, Streit WR (2009) Advances in recovery of novel biocatalysts from metagenomes. J Mol Microbiol Biotechnol 16:25–37
Yu EY, Kwon MA, Lee M, Oh JY, Choi JE, Lee JY, Song BK, Hahm DH, Song JK (2011) Isolation and characterization of cold-active family VIII esterases from an arctic soil metagenome. Appl Microbiol Biotechnol 90:573–581
Acknowledgments
This work was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Education, Science and Technology of Korea (Grant No. 2008-2004190).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Robb.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Choi, JE., Kwon, MA., Na, H.Y. et al. Isolation and characterization of a metagenome-derived thermoalkaliphilic esterase with high stability over a broad pH range. Extremophiles 17, 1013–1021 (2013). https://doi.org/10.1007/s00792-013-0583-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-013-0583-z