
Abstract
Objectives
To investigate the properties of a novel metagenome-derived member of the hormone-sensitive lipase family of lipolytic enzymes.
Results
A forest soil metagenome-derived gene encoding an esterase (Est06) belonging to the hormone-sensitive lipase family of lipolytic enzymes was subcloned, heterologously expressed and characterized. Est06 is a polypeptide of 295 amino acids with a molecular mass of 31 kDa. The deduced protein sequence shares 61% similarity with a hypothetical protein from the marine symbiont Candidatus Entotheonella sp. TSY1. Purified Est06 exhibited high affinity for acyl esters with short-chain fatty acids, and showed optimum activity with p-nitrophenyl valerate (C5). Maximum enzymatic activity was at 50 °C and pH 7. Est06 exhibited high stability at moderate temperatures by retaining all of its catalytic activity below 30 °C over 13 days. Additionally, Est06 displayed high stability between pH 5 and 9. Esterase activity was not inhibited by metal ions or detergents, although organic solvents decreased activity.
Conclusions
The combination of Est06 properties place it among novel biocatalysts that have potential for industrial use including low temperature applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metagenomics have provided a wealth of previously inaccessible genetic material from uncultivable organisms across a wide range of environments (de Castro et al. 2014). In addition to expanding the genetic diversity of known organisms, metagenomics has advanced the discovery of novel proteins, that can be used i.e., as catalysts for laundry, pharmaceutic or bioremediation applications (Selvin et al. 2012; Facchin et al. 2013). Esterases or carboxylesterases (EC 3.1.1.1), which catalyze synthesis and hydrolysis of short-chain glycerol esters, are an important group of industrially relevant biocatalysts. Several esterases have been isolated and identified by metagenomic approaches using environmental DNA from diverse environments ranging from bovine rumen (Rodríguez et al. 2015) to cryopegs in permafrost (Novototskaya-Vlasova et al. 2012). Esterases belong to a large group of lipolytic enzymes, that includes true lipases. A combination of cultivation dependent and independent approaches have expanded the original esterase classification of Arpigny and Jaeger (1999) from seven families to 15 families, the most recent being family XV (Charbonneau and Beauregard 2013).
A distinguishing feature of esterases is the presence of a GXSXG consensus sequence that contains the catalytic serine residue. This serine forms the nucleophilic part of a catalytic triad, together with aspartic acid or glutamine and histidine (Bornscheuer 2002; López-López et al. 2014). A substrate binds to the active site at the serine residue to form a tetrahedral intermediate that is stabilised by Asp/Glu and His residues. An acyl-enzyme complex is formed by elimination of alcohol, which is then hydrolysed by a water molecule to generate the final tetrahedral intermediate and free enzyme (Bornscheuer 2002). The diversity in primary amino acid sequences of esterases still results in a conserved tertiary structure that typically, consists of a N-terminal α-helical domain and a central barrel, catalytic domain of parallel β-strands known as the α/β hydrolase fold (Nardini and Dijkstra 1999; Lenfant et al. 2013).
As a group, esterases exhibit broad substrate range, temperature and pH spectra, and tolerance to organic solvents, metal ions and salts to varying degrees. Individually, they are highly selective enzymes, capable of chemo-, regio- and stereo-selective activity (López-López et al. 2014; Ma et al. 2014). Therefore, esterases and related lipolytic enzymes are important biocatalysts for organic synthesis, stereospecific drug production (Levisson et al. 2007), bioremediation of waste from oil refineries (Martínez-Martínez et al. 2014), detergent preparations (Zhang et al. 2014) and flavour development in the food industry (Mohamed et al. 2013).
In this study, a metagenome-derived gene encoding an esterase belonging to the hormone-sensitive lipase (HSL) family and the corresponding protein have been subcloned, heterologously produced and characterized. The gene was derived from a metagenomic plasmid (pLE06), which conferred lipolytic activity. The plasmid originated from a forest soil metagenome (Nacke et al. 2011). Lipolytic enzymes of the HSL group share sequence and structural homology to the catalytic domain of the mammalian hormone-sensitive lipase (Østerlund 2001). Subfamilies in this group possess either a GDSA or GTSA as a catalytic motif (Li et al. 2014), and were among the first esterases assigned to the α/β hydrolase superfamily (Lenfant et al. 2013).
Materials and methods
Bacterial strains and plasmids
The lipolytic recombinant strain E. coli/pLE06 harbors the metagenomic plasmid pLE06 including the putative esterase gene est06. The clone was derived from a functional screen of a metagenomic plasmid library, which was constructed from forest soil of Schorfheide-Chorin (Germany). Soil sampling, metagenomic library construction and identification of lipolytic clones was performed by Nacke et al. (2011). The plasmid pLE06 was used as template for amplification of est06. The corresponding nucleotide sequence is available at the National Center for Biotechnology Information (NCBI) gene bank under the accession number HQ156905. Escherichia coli strain BL21(DE3) and pET101/D (Invitrogen) were used as expression host and vector, respectively. Detailed maps of plasmids are provided in Supplementary Fig. 1.
Identification and analysis of est06 sequence
Amino acid sequences of esterases homologous to the deduced gene product of est06 were retrieved from the NCBI database and aligned using MUSCLE (Edgar 2004). Secondary and tertiary structure prediction was performed with I-TASSER (Zhang 2008; Roy et al. 2010; Yang and Zhang 2015) by modelling Est06 along the structure of E40 (Protein Data Bank code: 4XVC_A; Li et al. 2015), an esterase that shares 55% amino acid sequence similarity with Est06. Annotation of aligned sequences was performed with EsPript 3.0 (Robert and Gouet 2014). Additional parameters were calculated with the ProtParam tool at Expasy (Gasteiger et al. 2005).
Subcloning, overexpression and purification of Est06
The primer pair 5′-CACCATGTCGCAACAACAACTCC-3′ and 5′-CGCGGCGTTGGCGCGGACGAA-3′ were used to amplify the 888 bp fragment of est06 from pLE06. The est06 PCR product was purified by using agarose gel electrophoresis and the Qiaquick gel extraction kit, according to the manufacturer’s instructions (Qiagen). Subsequently, the purified product was cloned into expression vector pET101/D (Invitrogen) as recommended by the manufacturer. In this way, also sequences encoding a V5 epitope and a His6-tag were added to the 3′-end of the gene. The resulting recombinant vector (pEST06) was used to transform chemically-competent E. coli BL21 (DE3) (Invitrogen). Transformants were grown on lysogeny broth (LB) agar plates containing 15 g agar l−1, 100 mg ampicillin l−1.
For expression of est06 gene, single colonies were grown in 30 ml LB medium supplemented with 100 mg ampicillin l−1 overnight with shaking at 30 °C. Expression was initiated by inoculation of a 1 l-culture with 2% (v/v) of the overnight culture. At an OD600 of 0.4, expression was induced by the addition of 0.5 mM IPTG. Cells were harvested by centrifugation at 7000×g for 10 min at 4 °C after 6 h of incubation and washed twice in 50 mM sodium phosphate buffer (pH 7). The cells were suspended in LEW buffer (Macherey-Nagel, Düren, Germany) and lysed on ice by sonication with a UPS200S homogeniser (Hielscher Ultrasonics GmbH, Teltow, Germany).
Crude cell extract was cleared by centrifugation at 7000×g for 1 h. Purification of Est06 was initially carried out with the Protino-NI-TED packed columns as recommended by the manufacturer (Macherey-Nagel, Düren, Germany). Resulting elution fractions were pooled and dialysed overnight against 20 mM Tris/HCl buffer (pH 8) at 4 °C, and finally concentrated with the Vivaspin 10,000 Da exclusion columns (Sartorius AG). Further purification was performed by anion exchange chromatography by using a SOURCE 15Q 4.6/100 PE tricorn exchanger column and Äkta FPLC system (GE Healthcare Europe GmbH, Münich) over an elution gradient of 1 M NaCl/Tris/HCl (pH8). Active fractions were spectrophotometrically detected with p-nitrophenyl butyrate as substrate and, subsequently, pooled and concentrated with a Vivaspin column and 50 mM sodium phosphate (pH 7) as described above. The purity of Est06 was confirmed with SDS-PAGE. Protein concentrations were determined by the Bradford method.
Standard enzyme assays
The activity of Est06 was quantified by measuring initial rates of hydrolysis of p-nitrophenyl esters (pNP) and subsequent production of p-nitrophenol at 405 nm. Standard reactions were conducted at 25 °C in 1 ml containing 50 mM sodium phosphate buffer (pH 7) and 1 mM p-nitrophenyl valerate (C5) as substrate. Unless otherwise stated, the reaction was initiated by adding Est06 (2 U) to the assay reaction and monitored at 405 nm. The extinction coefficient of p-nitrophenol under the above-mentioned conditions was experimentally determined to be 8726 M−1 cm−1 (Hotta et al. 2002). One unit of enzyme activity was defined as the amount of enzyme required to release 1 µmol p-nitrophenol per min from p-nitrophenyl ester.
Characterisation of Est06
Esterase substrate specificity was assayed with triacylglycerides and p-nitrophenyl esters of fatty acids with different chain length. Plate assays for triacylglycerides were carried out by dropping 20 µl purified Est06 (7 U) on to LB agar plates supplemented with either 1% (w/v) tributyrin (C4), tricaproin (C6) or tricaprylin (C8), as well as 0.1% (w/v) tricaprin (C10), trilaurin (C12) or tripalmitin (C16). Zones of clearing on agar plates indicated lipolytic activity. Substrate specificity of Est06 for p-nitrophenyl esters was measured with 1 mM pNP acetate (C2), pNP butyrate (C4), pNP valerate (C5), pNP caproate (C6), pNP-caprylate (C8), pNP caprate (C10), pNP laurate (C12), pNP myristate (C14), or pNP palmitate (C16) under standard reaction conditions.
Optimum temperature was determined between 10 and 80 °C by using temperature-adjusted 50 mM sodium phosphate buffer (pH 7). Temperature stability of the enzyme was determined by incubating Est06 at 10, 25, 30, 40, 50 and 60 °C and measuring residual activity. As standard reaction conditions were defined for 25 °C, activity of Est06 at 25 °C was taken as 100%. The optimum pH of Est06 activity was assayed between pH 1 and 11, using the following overlapping buffer systems: KCl/HCl (pH 1–2.2), glycine/HCl (pH 2.2–3.6), sodium citrate (pH 3–6), sodium phosphate (pH 6–8), Tris/HCl (pH 7.6–9), CHES (pH 8.8–10), sodium carbonate (pH 9.6–10.6) and CAPS (pH 10–11). Due to pH-dependent absorption of p-nitrophenol, activity was monitored at the isosbestic point of p-nitrophenol (348 nm), the wavelength at which absorption is independent of pH (Hriscu et al. 2013). The pH stability of Est06 was assayed by incubating aliquots of enzyme in 20 µl buffer between pH 5 and 10 on a shaking incubator at 25 °C for 4 h. Aliquots of Est06 were pre-incubated between pH 5 and 10 in the respective buffers for 3 min. Residual enzyme activity was evaluated under standard reaction conditions and expressed as percentage of the activity of control reactions.
Effect of additives on Est06 activity
The effect of additives on Est06 activity was measured by pre-incubating the enzyme in assay buffer in 100 µl for 3 min at 25 °C on a shaking incubator. Est06 stability was determined by incubating the enzyme in assay buffer as above-mentioned for 1 h at 25 °C on a shaking incubator. Residual activity was calculated as a percentage of the activity of the control, which was not supplemented with the respective additive.
The effect of metal ions was studied by pre-incubating Est06 with metal ions at 1 and 5 mM in the assay buffer. The effect of salt on Est06 activity was measured directly by adding NaCl to the standard reaction assay from 0.05 to 3 M. Tolerance to salt was assayed by incubating the enzyme in 100 µl aliquots of assay buffers with NaCl from 0.05 to 3 M. The effect of detergents and inhibitors on Est06 activity was studied by pre-incubating Est06 with TritonX-100, Tween 20, Tween 80, SDS, 2-mercaptoethanol, dithiothreitol (DTT) or phenylmethylsulfonyl fluoride (PMSF) at 1 and 5 mM. The effect of organic solvents on Est06 activity was assayed by incubating the enzyme with 25 and 50% water-miscible organic solvents: acetone, 1-propanol, 2-propanol, ethanol, or dimethyl sulfoxide (DMSO) for 1 h.
Results
Identification and sequence analysis of a novel esterase-encoding gene
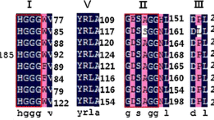
A lipolytic E. coli clone harbouring the recombinant plasmid pLE06 was recovered by functional screening of a forest soil metagenomic library (Nacke et al. 2011). Sequence analysis of the insert of the plasmid inserts revealed that it encodes a putative gene (est06) for a lipolytic enzyme, which is responsible for the lipolytic phenotype of the corresponding recombinant E. coli strain. The est06 gene (888 bp) encodes a polypeptide chain of 295 residues. Sequence similarity searches showed that Est06 shares amino acid sequence similarity of more than 50% with several hypothetical or uncharacterised esterases and lipases from uncultured prokaryotes (Supplementary Table 1), including an esterase (ETX01452.1) from Candidatus Entotheonella sp. (61%). Multiple sequence alignments of Est06 with other esterases revealed that Est06 belongs to the HSL group of lipolytic proteins (Fig. 1). These include a HGGG motif (residues 74 to 77) and a catalytic triad composed of Ser144, Glu238, and His268. Tertiary structure prediction confirmed the presence of two α-helices, α1 and α2, that constitute the N-terminal cap domain followed by the α/β fold of the catalytic domain (Supplementary Fig. 2). The hydrolase fold comprises eight β-strands that form parallel structures, with Ser144 located between β5 and α5, Glu238 between β7 and α7 and His268 between β8 and α9. Two cysteine residues, Cys46 and Cys170, which may participate in disulphide bridge formation are also present.

Multiple sequence alignment of Est06 and proteins related to the HSL family. Protein accession numbers are shown in parentheses. Catalytic triad residues are indicated by (filled triangle); residues of the oxyanion hole are marked with (filled circle), turns are marked with (T) and 310 turns are labelled with (η). Secondary structures are annotated on top of sequences and are modelled along E40, an HSL esterase that shares 55% sequence similarity with Est06. Regions with α-helices are shown by squiggles; regions with β-strands are marked by an arrow. Conserved residues are marked with a black background; similar residues are highlighted in bold, while sequences within the same group are marked with a border
Expression of the est06 gene and purification of the corresponding protein
Est06 was expressed in E. coli BL21 (DE3) for 6 h at 30 °C. The enzyme was purified to homogeneity by a combination of affinity and anion exchange chromatography, yielding 248 µg pure enzyme from 2 l culture, with a specific activity of (5.76 U/mg). The protein has a calculated molecular mass of approx. 31 kDa and a theoretical pI of 5.99. Separation of Est06 enzyme preparation by SDS-PAGE showed a single band of approx. 35 kDa (Fig. 2), which is in accordance with calculated mass including the V5 epitope and His6-tag added during cloning of the gene in the expression vector.

SDS-PAGE of purified, recombinant Est06. Lane 1 Marker PageRuler Prestained Protein Ladder (Fermentas), Lane 2 E. coli crude cellular lysate, Lane 3 purified Est06 after anion exchange chromatography
Substrate specificity of Est06
The ability of Est06 to hydrolyse different triacylglycerides and p-nitrophenyl esters of varying chain length was determined. A qualitative measure of triacylglyceride hydrolysis revealed the formation of halos on tributyrin-containing agar plates, which is indicative of lipolytic activity. However, none of the longer chain triacylglycerides were hydrolysed (data not shown), showing that Est06 could only hydrolyse short-chain triacylglycerides. Quantitative assays with p-nitrophenyl esters showed that Est06 exhibited a marked substrate preference for esters with short-chain fatty acids (Fig. 3). The maximum of enzyme activity was recorded with pNP valerate (C5) as substrate. Negligible activities were detected with substrates of longer chain fatty acids (>C6). The inability to hydrolyse long-chain glycerol esters or p-nitrophenyl esters demonstrated that Est06 is an esterase and not a lipase.

Substrate specificity of Est06. Activity was determined for pNP esters of varying chain length at 25 °C and pH 7. Relative activity was expressed as a percentage of pNP valerate (C5) enzyme activity (2.1 U/mg). Values are given as the mean of three experiments ± standard deviations
Optimum temperature and thermal stability of Est06
The maximum activity of purified Est06 toward pNP-valerate (C5) was at 50 °C under standard reaction conditions. Activity increased continually from 10 to 50 °C. At higher temperatures, activity decreased rapidly and was not detectable at 80 °C (Fig. 4a).

Biochemical characterisation of lipolytic enzyme Est06. a Effect of temperature on Est06 activity, b temperature stability profile of Est06, c effect of pH on Est06 enzyme activity, d stability of Est06 after incubation at 4 h at pH 5–10. Specific activities corresponding to 100% relative activity are: 3.7, 4.9, 1.1 and 1.4 U/mg for graphs A, B, C and D, respectively. Values are given as the mean of three experiments ± standard deviations
While Est06 is able to function above 50 °C, stability above this temperature was very low as activity was not recorded after 1 h at 50 °C and 60 °C. However, Est06 had remarkably high stability for extended incubation time (13 days end of experiment) at below 50 °C (Fig. 4b). Est06 retained all its activity at 10, 25 and 30 °C over the entire period. After six days of incubation at 40 °C, esterase activity remained above 70%. Considerable loss in Est06 activity (less than 65% residual activity) was observed only after the 10th day of incubation.
Optimum pH and pH stability of Est06
The optimum pH of Est06 was evaluated at 25 °C (see Fig. 4c). Activity was detected from pH 3.6–11. More than 70% activity was retained between pH 5 and pH 10. Maximum activity was at pH 7 and remained as high as 80% up to pH 10. Est06 retained more than 70% of its activity between pH 5 and 9 after 4 h incubation at 25 °C (Fig. 4d). Thus, Est06 is not only active over a wide range of pH values but is also stable over a broad pH spectrum and active under acidic as well as alkaline conditions.
Effect of additives on Est06 activity
Incubation of Est06 with different metal ions resulted in no significant loss of activity with the exception of Fe2+ (65% residual activity) and Co2+ (55% residual activity) (Fig. 5a). None of the metal ions significantly stimulated activity. The inability of EDTA to inhibit esterase activity is an indication that Est06 does not require metal ion activators or prosthetic groups for activity or stability (Mohamed et al. 2013; Peng et al. 2011). Addition of NaCl produced no stimulatory effect on Est06, and activity decreased linearly with increasing NaCl concentration (Fig. 5b). After incubation for 1 h, Est06 exhibited moderate stability with NaCl up to 2.5 M with the residual activity being over 75%. The results demonstrated that Est06 is not dependent on salt for activation or stability.

The effect of additives on Est06 activity and stability. Activity of Est06 under the influence of a metal ions, b salt and d detergents and inhibitors. Specific activity values expressed as percentages of the control reactions are: 4, 3.8, 5.2 and 3.2 U/mg for graphs A, B and C, respectively. Values are given as the mean of three experiments ± standard deviations
At low detergent concentrations (0.1%), Est06 retained most of its activity (above 90%), with the exception of SDS and PMSF, which resulted in loss of activity (Fig. 5c). At higher concentrations (1%), 2-mercaptoethanol and DTT exhibited a stimulatory effect on esterase activity (110%), while SDS and PMSF continued to inhibit Est06. Enzyme inhibition caused by SDS or PMSF is indicative of serine hydrolases (Peng et al. 2011). Tolerance of Est06 towards water-miscible organic solvents was generally low (Supplementary Table 2). An increase in enzyme activity (130%) was observed at 25% (v/v) DMSO although at a higher concentration (50%), DMSO decreased enzyme activity to 72%. All other tested organic solvents exhibited strong inhibition of Est06 activity, particularly at high solvent concentrations.
Discussion
Metagenomics is a powerful tool for the discovery of novel biocatalysts from various environments. In this study, a novel esterase from a forest soil metagenome was cloned, expressed and characterised. Est06 was identified through its ability to hydrolyse tributyrin on solid media and was initially classified as a putative esterase (Nacke et al. 2011). Further sequence analysis revealed that Est06 belongs to the HSL (Family IV) group of lipolytic proteins. The esterase shares similarity with a hypothethical protein from Candidatus Entotheonella sp. TSY1, which is a bacterial symbiont from deltaproteobacteria (Wilson et al. 2014). Esterases belonging to Family IV share amino acid sequence similarity to mammalian HSL, which regulates lipid metabolism in adipose tissue (Østerlund 2001). This indicates that bacterial and mammalian esterases share a common evolutionary root (Rhee et al. 2005). Signature motifs in this group include an HGGG motif, which is present in the Est06 protein sequence and participates in the formation of an oxyanion hole (Li et al. 2014). The oxyanion hole forms a pocket that aids in stabilising the negative charge of the tetrahedral intermediate during substrate binding (Lee et al. 2006). The catalytic triad in esterases/lipases is composed of Ser/His/Asp or, less commonly, Glu. Li et al. (2014) proposed two GXSXG motifs (GDSAG and GTSAG) within the HSL group and their separation into two distinct subfamilies. The active site serine in Est06 (Ser144) is located in the conserved GDSAG motif. The triad is completed by glutamine (Glu238) in the EXLLD motif and histidine (His268). Prediction of esterase tertiary structures has been achieved by using proteins sharing low amino acid sequence similarity. Berlemont et al. (2013) predicted the structure of organic-solvent tolerant esterase (RBest1) by using a protein that shared 36% amino acid sequence similarity, while Novototskaya-Vlasova et al. (2012) used as little as 16% similarity for their modelling of the cold-active lipase EstPc. In this study, tertiary structure prediction of Est06 was performed by using esterase E40 (Li et al. 2015), which shares 55% amino acid sequence similarity. Results confirm the α/β hydrolase fold reported for all members of lipase/esterase gene families (van Pouderoyen et al. 2001).
Esterases of the HSL family exhibit diverse optima for temperature and pH. Est06 was derived from a temperate forest soil metagenome, and as such it exhibits maximum catalytic activity under mesophilic conditions (below 50 °C). In addition, Est06 displays unprecedented thermal tolerance at moderate temperatures. Kovacic et al. (2016) demonstrated the ability for esterases to evolve thermal adaptations that resemble their bacterial host habitats. Thermal tolerance was attributed to the presence of loops in the tertiary fold that increases or decreases flexibility with changes in temperature. In comparison to the four esterases described by Kovacic et al. (2016), Est06 did not lose activity below 30 °C, a feature that highlights Est06 as a candidate catalyst in low temperature bioprocesses. Byun et al. (2007) reported for HSL member EstE1 that thermostability is imparted by hydrophobic interaction of valine and phenylalanine residues on β8, however these residues are absent in Est06 (Fig. 1). Additionally, factors such as hydrogen bonding and oligomerisation also play a role in esterase thermostability (De Simone et al. 2001; Rhee et al. 2006), which have not been evaluated in Est06. An adaptation to low working temperatures is an attractive trait for reactions in which high temperatures are not suitable, such as manufacture of thermolabile pharmaceutical products, food ingredients as well as production of cold-wash detergents (López-López et al. 2014). In addition, Est06 displays a broad pH spectrum for catalysis. Esterase activity was detected from as low as pH 3.6. The highest activities were observed from pH 5 to pH 10. Moreover, Est06 stability under different pH conditions was also high between pH 5 and pH 9. In comparison, a Geobacillus sp. TF17 esterase with comparable pH stability was described by Ayna et al. (2013), but this enzyme exhibited maximal activity in a much narrower pH range (pH 7–8). The Sulfolobus tokodaii esterase reported by Suzuki et al. (2004) was highly active between pH 5 and 9. In contrast, an esterase from Bacillus circulans was maximally active only between pH 8 and 9, and was stable between pH 6 to 8 (Kademi et al. 2000). Acid-stable enzymes such as Est06 are desirable catalysts in bioremediation processes for contaminated waste water or biofuel production sites, and application in lignocellulosic waste degradation (Sharma et al. 2012).
Est06 does not require metal ion activators or prosthetic groups for activity or stability (Peng et al. 2011), as addition of EDTA did not result in loss of enzyme activity. The activity of two HSL esterases (SBLip 2 and SBLip 5.1) identified by Biver and Vandenbol (2013) was similarly unaffected by the addition of EDTA. Although mechanisms for metal ion resistance in esterases are not well understood, Est06 was resistant to Cu2+ and Zn2+, metal ions usually with a strong inhibitory effect. Est06 retained more than 80% activity with both metal ions even at 5 mM. In contrast, an esterase (EstATII) characterised by Mohamed et al. (2013) was severely inhibited by Cu2+ and Zn2+. Such an ability to withstand additions of metal ions is an important factor for enzymes used in the bioremediation of environmental waste (Brault et al. 2012).
Serine hydrolases such as Est06 are commonly prone to inactivation by inhibitors such as PMSF and SDS. PMSF binds irreversibly to the active site serine residue and makes it inaccessible to substrate molecules (Sharma and Radha Kishan 2011). SDS inhibits enzyme activity by forming complexes with proteins through non-specific binding, which results in unfolding of the protein. Est06 was generally more stable in non-ionic detergents compared to SDS and PMSF. At 1% detergent, Est06 showed a decrease in residual activity with Tween 20 and Tween 80. Similarly, EstATII showed comparable behaviour towards Tween 20 and Tween 80 but with a severe decrease in activity (Mohamed et al. 2013).
Redox reagents, 2-mercaptoethanol and DTT exhibited minor effects on Est06 activity. Resistance to these reagents is related to the number of cysteine residues present in the protein, which are capable of forming disulphide bridges. Levisson et al. (2007) proposed that a single cysteine residue is unable to form disulphide bridges with nearby residues and as such, redox reagents such as 2-mercaptoethanol and DTT have no marked effect on activity. Therefore, the inability of 2-mercaptoethanol and DTT to inhibit or significantly activate Est06 confirmed that Cys46 and Cys170 form no disulphide bridges in Est06 tertiary structure. In contrast, Cys residues in esterase AprX-SK37 form 8 disulphide bridges, which subsequently contributed to strong inhibition by 2-mercaptoethanol and DTT (Phrommao et al. 2011). Neither activity nor stability of Est06 was dependent on NaCl. Halo-tolerance may be positively correlated with organic solvent tolerance (Berlemont et al. 2013). As salt causes a decrease in water molecules around the enzyme, these molecules would be replaced by organic solvents (Sellek and Chaudhuri 1999; Berlemont et al. 2013). A Salimicrobium sp. LY19 esterase supports these findings as it exhibited halo and organic solvent tolerant properties (Xin and Hui-Ying 2013). In comparison, low tolerance to salt by Est06 was accompanied by low organic solvent tolerance. Generally, enzymes have reduced activity in organic solvents than in water (Mattos and Ringe 2001). Est06 was inhibited by water-miscible organic solvents. At 25% total organic solvent concentration, there was a rough correlation between logPow, which is a measure of solvent polarity, and residual activity. Thus, residual activity decreased with increasing logPow (Supplementary Table 2). Esterase EstC2 showed similar inactivation by alcohols at 50% concentration (Kang et al. 2011). A 30% increase in activity was observed with 25% DMSO, although a higher concentration of DMSO (50%) had an inhibiting effect on Est06. Metin et al. (2006) attribute the activating effect of DMSO to the high rate of diffusion of substrate in its presence. Water-miscible solvents form a uniform phase with water and enable substrates quick access to the active site (Ogino and Ishikawa 2001). Mattos and Ringe (2001) counter argue that increased solvent polarity, as with DMSO, actually strips away the water layer around the enzyme and competes for hydrogen bonds in protein atoms, thus leading to inactivation. This was not the case with Est06 at low DMSO concentration but as concentration increased to 50%, a decrease in activity that could be caused by this inhibition phenomenon was recorded. In this study DMSO follows the amphiphilic effect described by Sellek and Chaudhuri (1999) in which DMSO functions as an activator at low solvent concentrations and an inhibitor at higher concentrations.
In conclusion, a temperate forest soil metagenome yielded an esterase with significant stability at moderate temperatures and broad pH range. Est06 shows no reliance on additives for activity or stability and exhibited marked resistance to metal ions, detergents and low concentrations of organic solvents. The combination of these properties demonstrate the robust capabilities of Est06 as a novel biocatalysts in low temperature applications.
References
Altschul SF, Madden TL, Schäffer AA et al (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acid Res 25:3389–3402
Arpigny JL, Jaeger K-E (1999) Bacterial lipolytic enzymes: classification and properties. Biochem J 343:177–183
Ayna Ç, Kolcuoğlu Y, Öz F et al (2013) Purification and characterization of a pH and heat stable esterase from Geobacillus sp. TF17. Turk J Biochem 38:329–336
Berlemont R, Spee O, Delsaute M et al (2013) Novel organic solvent-tolerant esterase isolated by metagenomics: insights into the lipase/esterase classification. Rev Argent Microbiol 45:3–12
Biver S, Vandenbol M (2013) Characterization of three new carboxylic ester hydrolases isolated by functional screening of a forest soil metagenomic library. J Ind Microbiol Biotechnol 40:191–200
Bornscheuer UT (2002) Microbial carboxyl esterases: classification, properties and application in biocatalysis. FEMS Microbiol Rev 26:73–81
Brault G, Shareck F, Hurtubise Y et al (2012) Isolation and characterization of EstC, a new cold-active esterase from Streptomyces coelicolor A3(2). PLoS ONE 7:e32041
Byun J-S, Rhee J-K, Kim ND et al (2007) Crystal structure of hyperthermophilic esterase EstE1 and the relationship between its dimerization and thermostability properties. BMC Struct Biol 7:47
Charbonneau DM, Beauregard M (2013) Role of key salt bridges in thermostability of G. thermodenitrificans EstGtA2: distinctive patterns within the new bacterial lipolytic enzyme Family XV. PLoS ONE 8:1–18
de Castro AP, Fernandes GdaR, Franco OL (2014) Insights into novel antimicrobial compounds and antibiotic resistance genes from soil metagenomes. Front Microbiol 5:1–9
De Simone G, Menchise V, Manco G et al (2001) The crystal structure of a hyper-thermophilic carboxylesterase from the archaeon Archaeoglobus fulgidus. J Mol Biol 314:507–518
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Facchin S, Diniz Alves PD, De Faria Siqueira F et al (2013) Biodiversity and secretion of enzymes with potential utility in wastewater treatment. Open J Ecol 3:34–47
Gasteiger E, Hoogland C, Gattiker A et al (2005) Protein identification and analysis tools on the ExPASy server. In: Walker JM (ed) The proteomics protocols handbook. Humana Press Inc., Totowa, pp 571–607
Hotta Y, Ezaki S, Atomi H, Imanaka T (2002) Extremely stable and versatile carboxylesterase from a hyperthermophilic archaeon. Appl Environ Microbiol 68:3925–3931
Hriscu M, Chiş L, Toşa M, Irimie FD (2013) pH-profiling of thermoactive lipases and esterases: caveats and further notes. Eur J Lipid Sci Technol 115:571–575
Kademi A, Aït-Abdelkader N, Fakhreddine L, Baratti JC (2000) Characterization of a new thermostable esterase from the moderate thermophilic bacterium Bacillus circulans. J Mol Catal B Enzym 10:395–401
Kang C-H, Oh K-H, Lee M-H et al (2011) A novel family VII esterase with industrial potential from compost metagenomic library. Microb Cell Fact 10:41
Kovacic F, Mandrysch A, Poojari C et al (2016) Structural features determining thermal adaptation of esterases. Protein Eng Des Sel 29:65–76
Lee M-H, Lee C-H, Oh T-K et al (2006) Isolation and characterization of a novel lipase from a metagenomic library of tidal flat sediments: evidence for a new family of bacterial lipases. Appl Environ Microbiol 72:7406–7409
Lenfant N, Hotelier T, Velluet E et al (2013) ESTHER, the database of the α/β-hydrolase fold superfamily of proteins: tools to explore diversity of functions. Nucleic Acid Res 41:D423–D429
Levisson M, van der Oost J, Kengen SWM (2007) Characterization and structural modeling of a new type of thermostable esterase from Thermotoga maritima. FEBS J 274:2832–2842
Li PY, Ji P, Li CY et al (2014) Structural basis for dimerization and catalysis of a novel sterase from the GTSAG motif subfamily of the bacterial hormone-sensitive lipase family. J Biol Chem 289:19031–19041
Li P-Y, Chen X-L, Ji P et al (2015) Interdomain hydrophobic interactions modulate the thermostability of microbial esterases from the hormone-sensitive lipase family. J Biol Chem 290:1118–11198
López-López O, Cerdán ME, González Siso MI (2014) New extremophilic lipases and esterases from metagenomics. Curr Protein Pept Sci 15:445–455
Ma B-D, Kong X-D, Yu H-L et al (2014) Increased catalyst productivity in α-hydroxy acids resolution by esterase mutation and substrate modification. ACS Catal 4:1026–1031
Martínez-Martínez M, Lores I, Peña-García C et al (2014) Biochemical studies on a versatile esterase that is most catalytically active with polyaromatic esters. Microb Biotechnol 7:184–191
Mattos C, Ringe D (2001) Proteins in organic solvents. Curr Opin Struct Biol 11:761–764
Metin K, Ateslier ZBB, Basbulbul G, Biyik HH (2006) Characterization of esterase activity in Geobacillus sp. HBB-4. J Basic Microbiol 46:400–409
Mohamed YM, Ghazy MA, Sayed A et al (2013) Isolation and characterization of a heavy metal-resistant, thermophilic esterase from a Red Sea brine pool. Sci Rep 3:3358
Nacke H, Will C, Herzog S et al (2011) Identification of novel lipolytic genes and gene families by screening of metagenomic libraries derived from soil samples of the German Biodiversity Exploratories. FEMS Microbiol Ecol 78:188–201
Nardini M, Dijkstra BW (1999) α/β hydrolase fold enzymes: the family keeps growing. Curr Opin Struct Biol 9:732–737
Novototskaya-Vlasova K, Petrovskaya L, Yakimov S, Gilichinsky D (2012) Cloning, purification, and characterization of a cold-adapted esterase produced by Psychrobacter cryohalolentis K5T from Siberian cryopeg. FEMS Microbiol Ecol 82:367–375
Ogino H, Ishikawa H (2001) Enzymes which are stable in the presence of organic solvents. J Biosci Bioeng 91:109–116
Østerlund T (2001) Structure-function relationships of hormone-sensitive lipase. Eur J Biochem 268:1899–1907
Peng Q, Zhang X, Shang M et al (2011) A novel esterase gene cloned from a metagenomic library from neritic sediments of the South China Sea. Microb Cell Fact 10:95
Phrommao E, Yongsawatdigul J, Rodtong S, Yamabhai M (2011) A novel subtilase with NaCl-activated and oxidant-stable activity from Virgibacillus sp. SK37. BMC Biotechnol 11:65
Rhee J-K, Ahn D-G, Kim Y-G, Oh J-W (2005) New thermophilic and thermostable esterase with sequence similarity to the hormone-sensitive lipase family, cloned from a metagenomic library. Appl Environ Microbiol 71:817–825
Rhee J-K, Kim D-Y, Ahn D-G et al (2006) Analysis of the thermostability determinants of hyperthermophilic esterase EstE1 based on its predicted three-dimensional structure. Appl Environ Microbiol 72:3021–3025
Robert X, Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324
Rodríguez MC, Loaces I, Amarelle V et al (2015) Est10: a novel alkaline esterase isolated from bovine rumen belonging to the new family XV of lipolytic enzymes. PLoS ONE 10:e0126651
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738
Sellek GA, Chaudhuri JB (1999) Biocatalysis in organic media using enzymes from extremophiles. Enzyme Microb Technol 25:471–482
Selvin J, Kennedy J, Lejon DPH et al (2012) Isolation identification and biochemical characterization of a novel halo-tolerant lipase from the metagenome of the marine sponge Haliclona simulans. Microb Cell Fact 11:72
Sharma A, Radha Kishan KV (2011) Serine protease inhibitor mediated peptide bond re-synthesis in diverse protein molecules. FEBS Lett 585:3465–3470
Sharma A, Kawarabayasi Y, Satyanarayana T (2012) Acidophilic bacteria and archaea: acid stable biocatalysts and their potential applications. Extremophiles 16:1–19
Suzuki Y, Miyamoto K, Ohta H (2004) A novel thermostable esterase from the thermoacidophilic archaeon Sulfolobus tokodaii strain 7. FEMS Microbiol Lett 236:97–102
van Pouderoyen G, Eggert T, Jaeger K-E, Dijkstra BW (2001) The crystal structure of Bacillus subtilis lipase: a minimal α/β hydrolase fold enzyme. J Mol Biol 309:215–226
Will C, Thürmer A, Wollherr A et al (2010) Horizon-specific bacterial community composition of german grassland soils, as revealed by pyrosequencing-based analysis of 16S rRNA genes. Appl Environ Microbiol 76:6751–6759
Wilson MC, Mori T, Rückert C et al (2014) An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 506:58–62
Xin L, Hui-Ying Y (2013) Purification and characterization of an extracellular esterase from a moderately halophilic bacterium. BMC Biotechnol 13:108
Yang J, Zhang Y (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43:W174–W181
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40
Zhang X-Y, Fan X, Qiu Y-J et al (2014) Newly identified thermostable esterase from Sulfobacillus acidophilus: properties and performance in phthalate ester degradation. Appl Environ Microbiol 80:6870–6878
Acknowledgements
We thank the DFG (German Science Foundation) for funding this work in the context of the graduate school ‘Die Bedeutung der Biodiversität für Stoffkreisläufe und biotische Interaktionen in temperaten Laubwäldern (GRK 1086)’. We thank Dr. Heiko Nacke for providing the lipolytic clone pLE06 used in this work, as well as Florian Jung and Jörn Lindemann for providing technical assistance.
Supporting information
Supplementary Table 1—Amino acid sequence similarities between Est06 and related lipolytic proteins.
Supplementary Table 2—The effect of water-miscible organic solvents on the stability of Est06.
Supplementary Figure 1—Maps of plasmids for cloning and expression of the lipolytic gene est06.
Supplementary Figure 2—Predicted tertiary structure of esterase Est06.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest exists.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dukunde, A., Schneider, D., Lu, M. et al. A novel, versatile family IV carboxylesterase exhibits high stability and activity in a broad pH spectrum. Biotechnol Lett 39, 577–587 (2017). https://doi.org/10.1007/s10529-016-2282-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2282-1