
Abstract
The OsmC-region (osmotically induced protein family) of the two-domain esterase EstO from the psychrotolerant bacterium Pseudoalteromonas arctica has been shown to increase thermolability. In an attempt to test if these properties can be conferred to another enzyme, we genetically fused osmC to the 3′-region of the family 8 xylanase encoding gene xyn8 from P. arctica. The chimeric open reading frame xyn8-OsmC was cloned and the chimeric protein was purified after heterologous expression in Escherichia coli. Xyn8 and Xyn8-OsmC showed cold-adapted properties (more than 60% activity at 0°C) using birchwood xylan as the preferred substrate. Maximal catalytic activity is slightly shifted from 15°C (Xyn8) to 20°C for Xyn8-OsmC. Thermostability of Xyn8-OsmC is significantly changed in comparison to wild-type Xyn8. The OsmC-fusion variant showed an apparent decrease in thermostability between 40 and 45°C, while both proteins are highly instable at 50°C.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Endo-1,4-β-xylanases (EC 3.2.1.8) are capable of xylan degradation by hydrolyzing β-1,4-glycosidic bonds of the polymeric substrate. During this process, xylo-oligosaccharides are produced, which are further hydrolyzed to xylose by β-xylosidase (Sunna et al. 1997). Based on the primary structure of the conserved catalytic region, xylanases are mainly grouped into glycosyl hydrolase (GH) families 10 and 11, but several enzymes capable of hydrolyzing β-1,4-xylosidic linkages were also found in other GH families, including family 8 xylanases (Henrissat 1991; Collins et al. 2005).
The vast majority of extensively characterized xylanases are of fungal or bacterial origin, and are described to be optimally active at mesophilic temperatures and neutral pH (Sunna and Antranikian 1997). These biocatalysts are used in a couple of industrial branches, such as baking, paper or animal feed industry (Sunna and Antranikian 1997; Collins et al. 2006). However, enzymes of microorganisms isolated from unusual biotopes promise attractive characteristics for industrial use. The characterization of xylanases from extremophiles has been mainly focused on the investigation of biocatalysts from thermo-, alkali- and acidophiles, while cold-adapted and halophilic enzymes have been only marginally examined (Waino and Ingvorsen 2003; Collins et al. 2005). Although the increase in temperature normally results in an enhanced reaction rate, it is often useful to run reactions at lower temperatures to preserve the properties of heat-labile products, e.g. in the food industry or to save energy (Collins et al. 2005).
Various strategies have been pursued in the past to alter catalytic characteristics of hydrolytic enzymes. Traditional strategies, such as directed evolution, gene shuffling or site-directed mutagenesis as well as timely synthetic biology approaches have already been used to improve enzymatic performance (Turner 2009; Liu and Schultz 2010; Hoesl et al. 2011). Furthermore, scientists have adopted nature’s strategy to create bi- or multifunctional enzymes by the fusion of genes, a process that also occurred during the course of evolution (Yourno et al. 1970; Khandeparker and Numan 2008). Moreover, molecular genetic approaches have been applied to replace a protein region by another resulting in a chimeric variant with novel features. In a recent study, the transfer of thermostable properties has been achieved by exchanging the N-terminal part of the mesophilic xylanase SoxB from Streptomyces olivaceovidiris against the analogous region of the heat-active xylanase TfxA of Thermomonospora fusca (Zhang et al. 2010).
In our previous study, we investigated the catalytic properties of the cold-active esterase EstO from Pseudoalteromonas arctica, which is composed of a C-terminal OsmC domain (osmotically induced protein family), in addition to the functional esterase domain at the N-terminus (Al Khudary et al. 2010). OsmC proteins are highly conserved among prokaryotes and were shown to be involved in mechanisms dealing with environmental stress (Rehse et al. 2004). The deletion of the OsmC domain dramatically increased the thermostability of the esterase and impaired the salt-mediated enzyme activation of EstO. In the present work, we demonstrate how to modify the biochemical properties of a cold-active family 8 xylanase from P. arctica by the genetic end-to-end fusion to the OsmC domain.
Materials and methods
Gene library construction and screening
P. arctica (DSMZ 18437) has been isolated at 4°C from sea ice samples in Spitsbergen, Norway (Al Khudary et al. 2008). A λ-phage gene library was created as described earlier and was used to screen for genes that encode xylanases (Al Khudary et al. 2010). A single clone, harboring plasmid pBK-CMV-Xyn8, generated dark blue zones after overnight incubation at 20°C on solid LB medium supplemented with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) and 50 μg/ml kanamycin, overlayed with AZCL-dye coupled xylan. Sequence of the plasmid pBK-CMV-Xyn8 was analyzed using standard primers T7 and T3 and primer walking was performed with oligonucleotides Xy2 (CGTAACAGTTTTTACTGTTG), Xy3 (GCAAACCCAGGTCAATGGTATG), Xy4 (GAGCGATTTGTAAAAGCAGGC) and Xy5 (CGATGGGTTATTATTAAATGC) to obtain the complete sequence of the insert.
Sequence analysis
BLAST analyses were performed using the public NCBI online database to obtain sequence data of related xylanases (Altschul et al. 1990). SignalP 3.0 online software was used to predict the signal sequence in Xyn8 (Emanuelsson et al. 2007).
Gene cloning of the xylanase
Cloning experiments were done under standard conditions, using Escherichia coli NovaBlue Singles (Novagen) for plasmid amplification and maintenance (Sambrook et al. 2001). The open reading frame xyn8 without the predicted secretion signal was amplified with primers Xy47SPBamHI-f (GGATCCGCATTTAATAATAACCCATCG, BamHI restriction site is underlined) and Xy47SalI-r (GTCGACTTAATTAAACGTGTTGTTATAAAAC, SalI is underlined) using genomic DNA as template. After subcloning into pDrive cloning vector (Qiagen) and sequence verification, the 1,224-bp BamHI/SalI digestion fragment was ligated into BamHI/SalI linearized expression vector pQE30 to give plasmid pQE-Xyn8. Similarly, a fusion variant of Xyn8 with C-terminal OsmC domain of P. arctica esterase EstO was cloned into pQE30 (Al Khudary et al. 2010). To obtain plasmid pQE-Xyn8-OsmC, fusion PCR was performed in three steps, amplifying secretion signal sequence less xyn8 with primer pair Xy47SPBamHI-f/Xy47SalI-r and estO OsmC domain with primers XO2-f (GTTTTATAACAACACGTTTAATGACAAAACTAAATACACGGCAAGTTTAA, part of xyn8 is given in regular letters, estO is given in italics) and XO2SalI-r (GTCGACTTATTCTACCAGTTCACTTACAATAACTGGG, SalI restriction site is underlined) using P. arctica genomic DNA as template. Subsequently, products of PCR 1 and 2 were used as templates for amplification of the full-length chimeric xyn8-OsmC product with primer pair Xy47SPBamHI-f/XO2SalI-r.
Expression and purification of the recombinant xylanase
Expression of xyn8 and xyn8-OsmC genes was performed using E. coli M15 [pREP4] strain harboring plasmids pQE-Xyn8 or pQE-Xyn8-OsmC, respectively. Transformants were grown at 37°C in 1.5-L batch fermentation cultures in liquid LB medium supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin. After OD600 = 0.6 was reached, expression of genes was induced with 0.1 mM IPTG at 15°C. Cells were harvested after 16 h and proteins were extracted from 1 g E. coli wet weight dissolved in 5 ml Lysis buffer (Qiagen, The QIAexpressionist) and cell disruption was achieved by sonication (Branson Sonifier, Branbury, USA). Protein purification was performed using a combination of Ni-NTA affinity chromatography and gel filtration according to the manufacturer’s recommendations. Active fractions were tested on 12% SDS-PAGE, pooled, concentrated and dialyzed against 120 mM universal buffer, pH 7.0 (Britton and Robinson 1931).
Protein activity assays
Protein concentrations in the eluted fractions were measured using the Bradford test (Bradford 1976). Enzymatic activity was quantified spectrophotometrically at 546 nm by measuring the release of reducing sugar using the DNS (3,5-dinitrosalicylic acid)-assay (Bailey 1988). All experiments were performed in triplicate. Enzyme activity in liquid solutions was determined for the following carbohydrate substrates: birchwood xylan, beechwood xylan, oat spelts xylan, carboxymethyl cellulose, Avicel, lichenin, laminarin, beta-glucan and starch. Reactions were performed at different temperatures (0–50°C) and different pH (pH 2–11). Stability tests were conducted after preincubation of the enzyme samples at various temperatures (at pH 7.0) or pH (at 4 or 20°C), prior to adding the enzyme samples to substrate solutions for activity measurement under optimal conditions (pH 7.0 and 15°C or pH 7.0 and 20°C, respectively). Determinations of the enzyme activities at temperature and pH values were conducted using 0.5% birchwood xylan as substrate.
Sequence accession number
The nucleotide sequence of P. arctica xyn8 was deposited in the EMBL database under accession number FR754551.
Results
P. arctica xyn8 encodes a family 8 xylanase
Xylanase Xyn8 from P. arctica has been identified in a xylanase specific gene library screen. Plasmid DNA from a single colony showing xylanase activity was isolated and an insert of 3,278 bps genomic DNA was sequenced. Plasmid pBK-CMV-Xyn8 contains the 1,281 bp xyn8 gene, which is highly similar to Pseudoalteromonas haloplanktis pXyl (96% identity at the DNA level and 98% identity at the protein level) and an upstream open reading frame encoding a hypothetical GAT_1-superfamily protein, which exhibits 62.8% identity at the protein level to a putative secreted protein from Saccharophagus degradans 2-40 (YP_526694.1). The C-terminal part of a third protein with 41.7% identity to the gluconolactonase domain protein from Teridinibacter turnerae T7901 (YP_003075103.1) is encoded downstream of the xylanase (Fig. 1a). The gene xyn8 has a total GC-content of 39.3% and the deduced peptide sequence encodes a protein of 426 amino acids with a predicted isoelectric point pI 8.46 and a molecular mass of 48.2 kDa (Table 1). A hypothetical Shine–Dalgarno sequence was identified at 7–12 nucleotides (AAGAGG) upstream of the ATG start codon and a putative signal peptide was predicted to be cleaved after Ala21 according to SignalP (Emanuelsson et al. 2007). The putative catalytic triad was found to be located at positions Glu99, Asp144 and Asp281, which is consistent to the situation in the related P. haloplanktis homologue (Van Petegem et al. 2003). Among the characterized family 8 xylanases, Xyn8 had sequence identities between 28.5 and 34.2% to the enzymes from the bacterial species Bacillus halodurans C-125, Bacillus sp. KK-1, Bifidobacterium adolescentis DSM20083 and from two uncultured microorganisms (Yoon et al. 1998; Brennan et al. 2004; Honda and Kitaoka 2004; van den Broek et al. 2005; Lee et al. 2006).

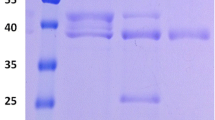
Xylanase Xyn8 from P. arctica. a Schematic representation of the genomic region. The open reading frame xyn8 encodes the P. arctica xylanase flanked by a gene encoding a putative GAT_1-superfamily protein (orf1) and another open reading frame, which encodes the C-terminal region of a protein similar to gluconolactonase domain proteins (orf3). The white box indicates the predicted signal peptide sequence. b Plate assays of xylanase activity. E. coli M15 [pREP4] clones transformed either with pQE-Xyn8 or pQE-Xyn8-OsmC were inoculated on LB medium containing 0.1 mM IPTG. After 24 h of growth, plates were overlayed with AZCL-dye coupled xylan. c SDS-PAGE analysis of the His-tagged xylanases Xyn8 and Xyn8-OsmC, purified by Ni-NTA affinity chromatography (Xyn8 and Xyn8-OsmC) and gel filtration (Xyn8). CE crude extract, Ni-NTA purification fraction after Ni-NTA affinity chromatography, GF gel filtration fraction
Expression and purification of Xyn8 and Xyn8-OsmC
The xyn8 gene was cloned into the vector pQE30, without the coding sequence of its predicted signal peptide, for further transformation in expression strain E. coli M15 [pREP4], thus enabling the production of a His-tagged variant. Fusion of the osmC domain from P. arctica esterase gene estO to xyn8 was done as described in “Materials and methods” and was also produced as an N-terminal His-tagged variant. E. coli cells expressing xyn8 or xyn8-OsmC were first tested on agar plates overlayed with AZCL-dye coupled xylan (Fig. 1b). Subsequently, cells were grown in liquid culture, harvested and disrupted by sonication and the lysates were assayed for xylanase activity. The best conditions for purification were examined and fermentation at 15°C enabled the production of high amounts of recombinant soluble xylanase (see in “Materials and methods”), while growth temperatures of 30 and 37°C led to the formation of inclusion bodies. The recombinant xylanases were purified from the culture supernatants of sonicated cells at 4°C using polypropylene columns loaded with Ni-NTA Agarose (Qiagen) in case of Xyn8-OsmC, while the ÄKTApurifier system with a Ni-NTA Agarose packed column followed by a gel filtration chromatography step at room temperature was used for Xyn8. Purification under room temperature conditions resulted in the degradation of Xyn8-OsmC, indicating that the fusion of the OsmC domain increased the thermolability of the full-length protein (data not shown). Xyn8 was purified 42.9-fold after the second step, with a specific activity of 291.8 U/mg, and yield of 2%, while Ni-NTA superflow column chromatography purification of Xyn8-OsmC (purification factor 6.0) resulted in a specific activity of 140.7 U/mg and a yield of 10.4% (Table 2). SDS-PAGE analyses revealed that purification rendered proteins with molecular masses of approximate 47 and 64 kDa (Fig. 1c), respectively. These results are consistent with the joined molecular masses of the full-length proteins (45.9 and 63 kDa) with the N-terminally fused His-tag peptide (1.3 kDa).
Biochemical characterization of Xyn8 and Xyn8-OsmC
To investigate the specificity of Xyn8 in comparison to Xyn8-OsmC, both variants were assayed with various substrates containing β-1,4-linked xylosepolysaccharide substrates. High hydrolytic activity was measured with all natural xylans tested, showing the maximum value on birchwood xylan (Table 3). The major products resulting from xylan hydrolysis were xylotriose and xylobiose, which indicated that Xyn8 is a strict endo-β-1,4-xylanase that only produces xylo-oligosaccharides with a low degree of polymerization (data not shown). Additionally, the identical results for Xyn8-OsmC clearly showed that the reaction mechanism is not influenced by the fusion of the OsmC domain. There was negligible activity against lichenin only for Xyn8-OsmC, but no activity against cellulose or other glucose-formed polysaccharide substrates (Table 3).
Enzymatic properties as a function of temperature and pH were determined using birchwood xylan as substrate (Fig. 2). The optimal temperature for activity was 15°C in case of Xyn8 and slightly shifted towards 20°C for Xyn8-OsmC (Fig. 2a). Xyn8 exhibited 61, 92 and 90% activity at 0, 10 and 30°C, while Xyn8-OsmC displayed 63, 69 and 78% activity. At 50°C both enzymes retained about 20% activity on birchwood xylan, but Xyn8 and Xyn8-OsmC completely lost their activities using beechwood xylan as substrate under these conditions (data not shown). Using birchwood xylan as substrate, more than 50% activity was measured between pH 5.0 and 9.0, with the optimum at pH 7.0 for both enzyme variants. No activity was detectable at pH 4.0 and 11.0 (Fig. 2b) and a sharp decrease of activity was measured at pH 10.0 (Xyn8, 46% remaining activity; Xyn8-OsmC, 38%).

Biochemical properties of Xyn8 and Xyn8-OsmC. a Temperature optimum. For the determination of the temperature optimum, activity assays were performed in universal buffer (pH 7.0) at a temperature range of 0–50°C. b pH optimum. The optimal pH for the recombinant enzymes was investigated by incubation of Xyn8 at 15°C in universal buffer (pH 2.0–11.0) or Xyn8-OsmC at 20°C. Activity of Xyn8 is given as filled squares, Xyn8-OsmC is indicated as filled triangles
Thermostability was tested by preincubation of the xylanase at 25–55°C for 30 min, 2 or 24 h, respectively, followed by an activity assay. Both enzymes were completely stable for 24 h at temperatures up to 37°C and fully inactive after 30 min at 50°C. After incubation at 40°C for 24 h, most of the activity of Xyn8-OsmC (25.4% remaining activity) was lost and Xyn8 was still 86% active (Table 4). Activity of Xyn8-OsmC drastically decreased after incubation for 30 min at 45°C (41.5% remaining activity), while Xyn8 retained 74.9% activity and 19.2% activity after 2 h. Prolonged incubation at 45°C resulted in the complete inactivation of Xyn8-OsmC.
Purified proteins were stable at 4°C and pH between 4.0 and 10.0, maintaining more than 70% activity for 24 h. At pH 3.0, Xyn8 exhibited 66% remaining activity after 30 min, while Xyn8-OsmC was more stable retaining 90%. This effect was actually more dramatic by incubation at 20°C and pH 4.0. Under these conditions, Xyn8-OsmC retained 89% activity for 24 h, while a total loss of activity (7.3%) has been observed for Xyn8. Additionally, no activity was measured for both proteins after 30 min incubation at 20°C and pH 3.0 (data not shown).
Since P. arctia optimally grows at 2–3% NaCl and the activity of esterase EstO was increased in the presence of 10 mM NaCl, but did not change for the OsmC deletion mutant EstOΔOsmC, the effect of NaCl on Xyn8 and Xyn8-OsmC was evaluated (Al Khudary et al. 2008, 2010). Xyn8 showed a high level of salt tolerance with at least 90% retained activity over a wide range of concentration (0.001–5.0 M NaCl). Furthermore, no loss of activity was observed for the Xyn8-OsmC variant after incubation in salt (data not shown). Moreover, the effect of the chelating agent EDTA on Xyn8 and Xyn8-OsmC was also investigated, because 10 mM EDTA completely inhibited the activity of EstO while 60% activity of EstOΔOsmC was retained in the presence of EDTA (Al Khudary et al. 2010). However, high concentrations of EDTA impaired the catalytic activity of Xyn8 and to a lesser extent of Xyn8-OsmC. Both enzyme variants were completely blocked in the presence of 50 mM EDTA, but Xyn8 retained 77, 69, 55 and 27% activity at 1, 5, 10 and 20 mM EDTA, respectively, while Xyn8-OsmC retained 89, 85, 70 and 34% activity, indicating that EDTA has minor influence on the Xyn8-OsmC activity.
Discussion
Most of the hyperthermophilic microorganisms belong to the Archaea, while the majority of cold-adapted species are of bacterial origin and cold environments are expected to provide new biocatalysts with unique characteristics (Egorova and Antranikian 2007). To investigate the biocatalytic properties of a cold-active xylanase, we expressed the xyn8 gene from P. arctica in E. coli. The aerobic, marine bacterium P. arctica grows optimally at pH 7.0–8.0 and 10–15°C in media supplemented with 2–3% NaCl (Al Khudary et al. 2008). Xyn8 is clearly homologous to the well-known pXyl from the close relative Antarctic bacterium P. haloplanktis, which has been classified as a GH family 8 member (Collins et al. 2002). The residues of the catalytic triad were predicted to act as the general acid (Glu78) and base (Asp302) catalysts. Asp165 plays a role in sugar ring distortion and transition state stabilization (Collins et al. 2005). Alanine mutants were examined in B. halodurans Rex, and proved essential in family 8 xylanases (Honda and Kitaoka 2004). All these residues are conserved in Xyn8.
Activity measurements revealed that P. arctica Xyn8 was most active between 10 and 40°C with maximal activity at 15°C, and functional between pH 5 and 10. Moreover, the enzyme was pH and temperature stable. Furthermore, Xyn8 hydrolyzes all the xylans tested, but no activity was found against substrates with a glucose backbone (carboxymethyl cellulose, Avicel, lichenan, laminarin, barley glucan and starch). Proteins from extremophilic organisms might have undergone adaptation and optimization processes during the course of evolution, making them amazingly efficient under harsh conditions. Besides sequence-specific mutagenesis after gene duplication processes (neofunctionalization), fusion of two proteins with different characteristics has also been discussed to be an evolutionary force to gain new functions or lose others (Chandrasekaran and Betrán 2008). Keeping this possibility in mind, we designed a chimeric protein composed of Xyn8 and the OsmC domain of the functional esterase EstO from P. arctica fused to the C-terminus to modify the catalytic properties of the xylanase (Al Khudary et al. 2010). An internal linker, which separates the esterase domain from OsmC in EstO was also maintained in the Xyn8-OsmC construct to assure the retention of the structure. The fusion of the OsmC domain slightly increases the pH stability under acidic conditions, but more important the temperature-dependent stability was drastically reduced, which is in good agreement with the catalytic properties of EstO and the truncated EstOΔOsmC variant (Al Khudary et al. 2010). Additionally, due to the instability of the Xyn8-OsmC variant, it was not possible to purify the enzyme at room temperature, indicating that the increased heat-sensitivity is specifically mediated by the OsmC-fusion.
Another effect that has been described for EstO, but not for EstOΔOsmC, was the stimulation of the activity by NaCl (10 mM increased the activity by 30%). It was proposed that the OsmC domain of the P. arctica esterase EstO is susceptible to conformation modification by NaCl (Al Khudary et al. 2010). However, the fused OsmC domain did not influence the activity of Xyn8 at all with regard to salt-mediated activation. In this context, it has to be discussed, that a possible effect might be overlayed by the remarkable salt tolerance of Xyn8. The enzyme is only slightly affected by the addition of NaCl up to a concentration of 5 M. Nevertheless, beside xylanases from halotolerant species, only a single enzyme from the marine bacterium Glaciecola mesophila KMM 241 was shown to be equally salt-tolerant (Waino and Ingvorsen 2003; Guo et al. 2009).
The control of enzymatic activity by heat inactivation is of great interest in industries where low temperatures are required (e.g. food industry) and it is a challenge to obtain more flexible enzymes for various applications.
References
Al Khudary R, Stösser NI, Qoura F, Antranikian G (2008) Pseudoalteromonas arctica sp. nov., an aerobic, psychrotolerant, marine bacterium isolated from Spitzbergen. Int J Syst Evol Microbiol 58:2018–2024
Al Khudary R, Venkatachalam R, Katzer M, Elleuche S, Antranikian G (2010) A cold-adapted esterase of a novel marine isolate, Pseudoalteromonas arctica: gene cloning, enzyme purification and characterization. Extremophiles 14:273–285
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Bailey MJ (1988) A note on the use of dinitrosalicylic acid for determining the products of enzymatic reactions. Appl Microbiol Biotechnol 29:494–496
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brennan Y et al (2004) Unusual microbial xylanases from insect guts. Appl Environ Microbiol 70:3609–3617
Britton HTK, Robinson RA (1931) CXCVIII—universal buffer solutions and the dissociation constant of veronal. J Chem Soc 458:1456–1462
Chandrasekaran C, Betrán E (2008) Origins of new genes and pseudogenes. Nat Educ 1
Collins T, Meuwis MA, Stals I, Claeyssens M, Feller G, Gerday C (2002) A novel family 8 xylanase, functional and physicochemical characterization. J Biol Chem 277:35133–35139
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29:3–23
Collins T et al (2006) Use of glycoside hydrolase family 8 xylanases in baking. J Cereal Sci 43:79–84
Egorova K, Antranikian G (2007) Biotechnology. In: Garret RA, Klenk H-P (eds) Archaea—evolution, physiology and molecular biology. Blackwell, USA, pp 295–321
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2:953–971
Guo B, Chen XL, Sun CY, Zhou BC, Zhang YZ (2009) Gene cloning, expression and characterization of a new cold-active and salt-tolerant endo-beta-1,4-xylanase from marine Glaciecola mesophila KMM 241. Appl Microbiol Biotechnol 84:1107–1115
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Hoesl MG et al (2011) Lipase congeners designed by genetic code engineering. ChemCatChem 3:213–221
Honda Y, Kitaoka M (2004) A family 8 glycoside hydrolase from Bacillus halodurans C-125 (BH2105) is a reducing end xylose-releasing exo-oligoxylanase. J Biol Chem 279:55097–55103
Khandeparker R, Numan MT (2008) Bifunctional xylanases and their potential use in biotechnology. J Ind Microbiol Biotechnol 35:635–644
Lee CC, Kibblewhite-Accinelli RE, Wagschal K, Robertson GH, Wong DW (2006) Cloning and characterization of a cold-active xylanase enzyme from an environmental DNA library. Extremophiles 10:295–300
Liu CC, Schultz PG (2010) Adding new chemistries to the genetic code. Annu Rev Biochem 79:413–444
Rehse PH, Ohshima N, Nodake Y, Tahirov TH (2004) Crystallographic structure and biochemical analysis of the Thermus thermophilus osmotically inducible protein C. J Mol Biol 338:959–968
Sambrook J, Fritsch E, Maniatis T (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor, New York
Sunna A, Antranikian G (1997) Xylanolytic enzymes from fungi and bacteria. Crit Rev Biotechnol 17:39–67
Sunna A, Moracci M, Rossi M, Antranikian G (1997) Glycosyl hydrolases from hyperthermophiles. Extremophiles 1:2–13
Turner NJ (2009) Directed evolution drives the next generation of biocatalysts. Nat Chem Biol 5:567–573
van den Broek LA, Lloyd RM, Beldman G, Verdoes JC, McCleary BV, Voragen AG (2005) Cloning and characterization of arabinoxylan arabinofuranohydrolase-D3 (AXHd3) from Bifidobacterium adolescentis DSM20083. Appl Microbiol Biotechnol 67:641–647
Van Petegem F, Collins T, Meuwis MA, Gerday C, Feller G, Van Beeumen J (2003) The structure of a cold-adapted family 8 xylanase at 1.3 Å resolution. Structural adaptations to cold and investigation of the active site. J Biol Chem 278:7531–7539
Waino M, Ingvorsen K (2003) Production of beta-xylanase and beta-xylosidase by the extremely halophilic archaeon Halorhabdus utahensis. Extremophiles 7:87–93
Yoon KH, Yun HN, Jung KH (1998) Molecular cloning of a Bacillus sp. KK-1 xylanase gene and characterization of the gene product. Biochem Mol Biol Int 45:337–347
Yourno J, Kohno T, Roth JR (1970) Enzyme evolution: generation of a bifunctional enzyme by fusion of adjacent genes. Nature 228:820–824
Zhang S, Zhang K, Chen X, Chu X, Sun F, Dong Z (2010) Five mutations in N-terminus confer thermostability on mesophilic xylanase. Biochem Biophys Res Commun 395:200–206
Acknowledgments
The authors thank Rami Al Khudary and Nele Stösser for the identification of pBK-CMV-Xyn8. Torben Rehn and Ute Lorenz are thanked for the help with some experiments and Mazen Rizk for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Santos.
Rights and permissions
About this article
Cite this article
Elleuche, S., Piascheck, H. & Antranikian, G. Fusion of the OsmC domain from esterase EstO confers thermolability to the cold-active xylanase Xyn8 from Pseudoalteromonas arctica . Extremophiles 15, 311–317 (2011). https://doi.org/10.1007/s00792-011-0361-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-011-0361-8