
Abstract
The sequence of the genome of the first alkaliphilic bacteriophage has been determined. Temperate phage BCJA1 possesses a terminally redundant genome of approximately 41 kb, with a mol% G+C content of 41.7 and 59 genes arranged predominantly into two divergent transcriptons. The integrase gene of this phage is unique in that it contains a ribosomal slippage site. While this type of translational regulation occurs in the synthesis of transposase, this is the first time that it has been observed in a bacteriophage integrase. The DNA replication, recombination, packaging, and morphogenesis proteins show their greatest sequence similarity to phages and prophages from the genus Streptococcus. Host specificity, lysin, and lysogeny maintenance functions are most closely related to genes from Bacillus species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alkaliphilic microorganisms, which grow optimally at pH values between 9 and 11, are widespread in nature. They have been isolated from many naturally occurring alkaline environments (such as soda lakes) man-made alkaline environments (e.g., bauxite processing wastes), as well as many other sites (e.g., soils and feces). Both bacteria and archaea have alkaliphilic representatives. Curiously, in spite of the widespread distribution of alkaliphilic microorganisms, reports of viruses active against these organisms are extremely rare. Indeed, isolation of viruses active against most extremophilic microorganisms, with the exception of thermophilic and hyperthermophilic bacteria and archaea (Rachel et al. 2002; Prangishvili 2003), is uncommon. Recently, the first complete sequence of a virus, ϕCh1, infecting the haloalkaliphilic archaeon Natrialba magadii, was reported (Klein et al. 2002). The sequence of the halotolerant alkaliphilic bacterium Oceanobacillus iheyensis (Takami et al. 2002) contains a defective prophage, called Bha35X (Mantri and Williams 2004a, 2004b) or Bh1 (Canchaya et al. 2003). Two phages of alkaliphilic bacilli have been reported, phage A1-K-I active against a not further characterized Bacillus sp. (Horikoshi and Yonezawa 1978) and, more recently, the temperate bacteriophage BCJA1, which infects the obligately alkaliphilic species Bacillus clarkii (Jarrell et al. 1997).
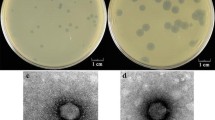
Bacteriophage BCJA1c is a member of the Siphoviridae family with B1 morphology. It possesses an isometric head that measures 65 nm in diameter and a noncontractile tail of 195 nm in length. Analysis of the protein composition of the phage revealed approximately ten structural proteins, with the major protein species having apparent molecular masses of 36.5 and 28 kDa. The latter were considered as possibly the major head and tail proteins, respectively (Jarrell et al. 1997). The genome was estimated to be between 32.1 and 34.8 kb in length, with a mol% G+C content of 45.6. This contribution represents the first complete genome sequence of a bacteriophage active against an obligately alkaliphilic bacterium.
Materials and methods
Organism and growth medium
Bacillus clarkii JaD is an obligate alkaliphile isolated from alkaline red mud from bauxite processing waste (Agnew et al. 1995). It was grown at 37°C in the growth medium (pH 10) recommended for Bacillus alcalophilus (Slepecky and Hemphill 1992). The clear plaque mutant of bacteriophage BCJA1, BCJA1c, was used in this study as it routinely grew to a higher titer than the wild-type version of the bacteriophage (Jarrell et al. 1997). Bacteriophage BCJA1c (accession number HER 428) and host B. clarkii (accession number HER 1406) have been deposited in the Felix d’Herelle Reference Centre for Bacterial Viruses, Faculty of Science, Laval University, Que., Canada.
Isolation of bacteriophage BCJA1c DNA
DNA was isolated from CsCl-purified bacteriophage BCJA1c as previously described (Jarrell et al. 1997).
Sequencing procedure
A combination of three procedures was used to determine the sequence of BCJA1c DNA. In the first case, the DNA was partially digested with a mixture of four restriction endonucleases that recognize 4-bp sequences and produce blunt-ended fragments. These were AccII (Amersham Biosciences, Baie d’Urfé, Que., Canada), HaeIII, AluI (New England Biolabs, Beverly, Mass., USA), and HpyF44III (MBI Fermentas, Burlington, Ont., Canada). After preparative agarose gel electrophoresis, fragments of 1.5–3 kb were recovered using Prep-A-Gene matrix (Bio-Rad Laboratories, Philadelphia, Penn., USA) and ligated into pUC18 digested with SmaI and dephosphorylated with bacterial alkaline phosphatase. The constructs were electroporated into Escherichia coli DH5α [F− ϕ80dlacZM15 (lacZYA-argF) U169 recA1 endA1 hsdR17(r − k , m + k ) phoA supE44 λ− thi-1 gyrA96 relA1], and colonies were selected in Luria agar (Difco) containing ampicillin (100 μg/ml, Sigma-Aldrich Canada, Oakville, Ont., Canada) and 40 μg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside). Individual clones were grown in Terrific Broth (Difco), and plasmid DNA was isolated using the alkaline lysis procedure (Sambrook et al. 1989). The DNA inserts were sequenced at the McGill University and Genome Québec Innovation Centre (Montreal, Que., Canada). Gap closure was accomplished using primer walking off the phage DNA at the Robarts Research Institute (London, Ont., Canada) and by PCR amplification with specific primers and amplicon sequencing at the Centre for Applied Genomics (Toronto, Ont., Canada). The sequence was stripped of poor quality and vector data, using SeqMan (DNASTAR, Madison, Wis., USA) and assembled into contigs.
Sequence analysis
Open reading frames were identified using Kodon (Applied Maths, Austin, Tex.). Protein masses and isoelectric points were calculated using EditSeq (DNASTAR). Potential homologues were identified using BLASTP (Altschul et al. 1990) or PSI-BLAST (Altschul et al. 1997) at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov). Where homologues were identified, pairs of sequences were compared using the Institute of Human Genetics’ program ALIGN at its Web site (http://www2.igh.cnrs.fr/bin/align-guess.cgi). Conserved protein motifs were identified as part of BLASTP analyses include Pfam (Bateman et al. 2002), Smart (Letunic et al. 2002; Schultz et al. 2000; Hofmann et al. 1999), CDD (Marchler-Bauer et al. 2003), and COG (Tatusov et al. 2003) databases. To predict transmembrane domains, TMHMM (Sonnhammer et al. 1998) at the Center for Biological Sequence Analysis at the Technical University of Denmark (http://www.cbs.dtu.dk/services/TMHMM-2.0/) was employed. Helix–turn–helix motifs were identified using the Pôle Bio-Informatique Lyonnais Network Protein Sequence Analysis server at http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_hth. html.
Potential integration host factor (IHF)-binding sites were assessed using PromScan (Studholme and Dixon, 2003) at http://molbiol-tools.ca/promscan/, while potential transcriptional terminators (Brendel et al. 1986; Brendel and Trifonov 1984) were assessed using GeSTer (Unniraman et al. 2002). Promoters were predicted using Softberry’s BPROM program at http://www.softberry.com/berry. phtml?topic=promoter. Repeat sequences were found using PHIRE (Lavigne et al. 2004).
Nucleotide sequence accession number
The BCJA1c sequence has been deposited with GenBank (accession number AY616446).
Results
DNA sequence analysis
Based upon restriction analysis the predicted size of BCJA1 DNA was 32.1–34.8 kb (Jarrell et al. 1997). DNA sequencing indicates that the unique sequence is 41,092 bp, and that this phage possesses terminal repetitious sequences of about 0.35 kb. For ease of presentation, the nonredundant genomic sequence was opened adjacent to a 33-bp bidirectional ρ-independent terminator (AAAAAAAGAGCCCGGTTAATTCCGGGCTTTTTT) with a calculated ΔG of −7.3 kcal/mol located downstream of gene 59 (lysin).
The overall base composition of the viral DNA (41.7 mol% G+C) was almost 4% lower than the published value (45.6%) determined from the melting profile (Jarrell et al. 1997). The current value is very similar to that of the host bacterium, Bacillus clarkii (42.7 mol% G+C, Nielsen et al. 2004), which is a not unexpected observation, since the base composition of temperate phage genomes usually closely match that of their hosts (A.M. Kropinski, unpublished results).
Description of selected protein coding sequences
Four criteria were used to define potential coding sequences (CDSs): either they had to exhibit sequence similarity to existing genes in the databases or they had to (1) contain >30 codons, (2) be preceded by a sequence displaying similarity to the consensus ribosome-binding site TAAGGAGGT (Shine and Dalgarno 1974, 1975), and (3) usually employ ATG or GTG as initiation codons. As with other phage genomes, the genes of BCJA1c were densely packed with many incidences of overlapping gene sequences.
A total of 59 potential CDSs were discovered, of which 68% encoded polypeptides that showed significant sequence similarity to proteins in the GenBank databases (Table 1; Fig. 1). In approximately 40% of the cases where homologues exist, they are to uncharacterized or hypothetical proteins. The properties of some of the CDSs that had identified function are discussed in the following sections, with special emphasis to the genes involved in replication, morphogenesis, integration, lysis, and regulation.

Genome map of phage BCJA1c with integrase (black), regulatory genes (red), genes involved in replication or recombination (dark blue), DNA packaging (green), morphogenesis (brown), and lysis (pink). Coding sequences with undefined homologues are in light blue, while unique genes are displayed in outline. ρ-Independent terminators are shown as ball-on-stick figures. Regions of homology to Streptococcus pyogenes prophage 370.1 are shown as filled purple bars, while regions with similarity to Bacillus halodurans proteins are illustrated by purple boxes. With the exception of the fourth line, each line in the diagram represents 11.2 kb
Replication and recombination
The replication of this phage has not been studied, but it probably involves genomic circularization mediated through recombination between the redundant ends of the molecule, followed by expansion involving θ-type replication and ultimately rolling circle, or σ replication, providing the substrate for packaging. Bacteriophage genomes often contain genes with homology to primases or helicases, and BCJA1 is no exception. Gene 16 specifies an ATP-dependent DNA helicase containing the NTP-binding domain T31 GCGKT36 [TGxGK(T/S), Walker box A, Walker et al. 1982] and a D122 EAH125 motif involved in Mg2+ binding and catalysis (Walker box B). What sets BCJA1 apart from most phages is the presence of a RepA homologue. This protein, often associated with plasmid replication, is involved in binding to the origin of replication and recruiting replication proteins. Only two other phages possess RepA proteins, and these are the Escherichia coli plasmid prophages of N15 (Ravin et al. 2003) and P1 (Chattoraj et al. 1985; Abeles et al. 1984). This suggests that in the lysogenic state BCJA1 also exists extrachromosomally.
RepA binds to iterated sequences (iterons) on N15 and P1 DNA, and these are characteristic of many phage replication origins. The use of base compositional skew analysis has been used to define replication origins (Ori) (Lobry 1999; Kowalczuk et al. 2001; Grigoriev 1998), but unfortunately, in this case it did not prove informative. In addition to iterons, Ori regions contain protein-binding motifs for replication proteins such as DnaA, HU, FIS, and IHF, the latter three of which induce DNA bending (Betermier et al. 1994; Grove et al. 1996; Swinger et al. 2003). A sequence (TTTTCCACA) was found on the minus strand centered at 10780, i.e., within gene 17, which is identical to the consensus DnaA-binding site (TTWTNCACA) (http://www.nmr.chem.uu.nl/~mike/ html/dna.html). There are two strong IHF-binding sites in CDS 16. Numerous direct repeats lie in the CDS 17–22 region including two copies of ATTCGAAGCAT, three copies of GGCAAAAAG, and four copies of TTGAAGGA. These all lie within approximately 4 kb of each other. A site (TCACAGAATTACTCAACAAAAAAGGA) that bears strong sequence similarity to the consensus FIS-binding site (TN2YN2AAWTN7AAWWRA) is found between 9622 and 9647. These finding suggest that the BCJA1 Ori lies somewhere between CDSs 17 and 22.
Other CDSs which may participate in DNA replication or recombination include RecF (CDS 13) and RusA endonuclease (Holliday junction resolvase) homologues. The latter is sufficiently similar to the E. coli enzyme to permit molecular modeling.
Lysis
Bacteriophage-induced lysis usually involves a two-gene lysis cassette composed of a holin and an endolysin (murein hydrolase). The holin creates pores in the inner or cytoplasmic membrane, permitting the endolysin to access the peptidoglycan layer in the periplasm, resulting in cell lysis and release of progeny viruses (Young and Bläsi 1995; Young 1992). With some exceptions the endolysin gene is preceded or overlapped by a gene encoding a holin. The product deduced from CDS 59 is a polypeptide of 355 amino acids that displays sequence similarity to a variety of putative bacterial or prophage lytic proteins classified at N-acetylmuramoyl-L-alanine amidases. This enzyme rather than lysozyme is also used by Streptococcus pneumoniae prophages MM1 (Obregon et al. 2003) and EJ-1 (NC_005294), Bacillus subtilis phages SPβc2 (NC_001884) and SPP1 (Alonso et al. 1997), and Bacillus cereus prophage phBC6A51 (Ivanova et al. 2003).
Holins are characterized by their relatively small size (71–161 amino acid residues), contain two to three membrane-spanning helices and a charged C terminus, and exhibit poor sequence identity to other members of this group of functionally similar proteins (Young 1992; Grundling et al. 2000; Young and Bläsi 1995). The predicted product of CDS 57 is likely the holin, since it is a protein containing 87 amino acids arranged into two transmembrane domains with a high concentration of basic amino acids at the C terminus.
Integration
In the lysogenic state, prophage genomes are mostly found integrated into the host genome. Integration is brought about through site-specific recombination between homologous phage (attP) and bacterial (attB) sites, catalyzed by the phage protein integrase (Int) in conjunction with the host-encoded accessory protein IHF (Campbell 1992). In the case of BCJA1, two proteins show homology to integrases. These are the products of CDSs 3A and 3B, respectively. Since BLASTX analysis of this region of the genome showed that a potential mistake had been made in the sequencing, this region was amplified by PCR and resequenced. However, the sequence proved to be correct. Interestingly, CDS 3B contains a potential ribosome slippage site (Pande et al. 1995; Alam et al. 1999; Harger et al. 2002), which, if utilized, would result in the synthesis of a fusion protein with 369 amino acid residues containing the N terminus of CDS 3B and the C terminus of CDS 3A. Immediately downstream of this site is a stem-loop structure that would result in ribosomal pausing, but it lacks the characteristic pseudoknot structure (Alam et al. 1999; Reeder and Giegerich 2004). While this type of translational regulation occurs in the synthesis of transposase (Gertman et al. 1986), this is the first time that it has been observed in a bacteriophage integrase.
The Int family of recombinases (tyrosine recombinases) has been the subject of considerable research activity, revealing which residues are conserved and which are involved in attP binding, dimerization, and catalysis (Nunes-Duby et al. 1998; Esposito and Scocca 1997; Bankhead and Segall 2000). While these proteins show a poor level of overall sequence similarity, motif analysis against the the CDD database (Marchler-Bauer et al. 2003) revealed the presence of cd01189. This motif is defined as INT_phiLC3_C (phiLC3 phage and phage-related integrases, site-specific recombinases), which contains three conserved oligopeptides, I223 NKTW227, H309 GLRHTHAS317, and Y328 VSERLGHADI338. The active site lies within the tetrad E344 YAH347. The closest homologues to the BCJA1c integrase are to be found in Streptococcus thermophilus bacteriophages Sfi21 (Brüssow and Bruttin 1995) and ϕO1205 (Stanley et al. 1997).
The attP sites contain regions for the binding of Xis, Fis, and IHF proteins. While we found no evidence for a Xis homologue, examination of the region downstream of gene 3 resulted in the identification of two pairs of the sequence TTTTACACA within a 228-bp region, which we propose may represent arm-type integrase-binding sites (Nash 1990). They overlap with two IHF-binding sites bearing strong sequence similarity to the consensus and two potential Fis sites. Finally, attP regions are usually AT-rich, and the 282-bp downstream of int has an average 67% A+T content.
Immunity region
BCJA1 is a temperate phage, and our analysis has revealed that central regulation probably involves, as in coliphage λ, opposing repressor (CDS 5) and antirepressor (cro, CDS 6) genes. Both contain helix–turn–helix motifs associated with DNA binding.
We currently do not know whether the wild-type phage BCJA1 is UV inducible; however, this is doubtful, because the repressor protein lacks both Ala–Gly or Cys–Gly motifs that are associated with RecA-stimulated autodigestion of the repressor proteins in phages such as λ, ϕ80, and P22 (Little 1991; Craig and Roberts 1980; Roberts et al. 1977; Raymond-Denise and Guillen 1991). It also lacks a C-terminal protease domain associated with repressor cleavage and induction.
If CDS 6 encodes the repressor, we would expect that repressor-binding sites might be located nearby. Indeed, we identified two 16-bp hyphenated inverted repeats with the half consensus sequence AGCTAA in the CDS 5–6 intergenic region. In almost all cases, phage operators are 14 [e.g., Mu (Goosen and van de Putte 1987)]- to19-bp [e.g., ϕ80, (Ogawa et al. 1988)] hyphenated palindromes. Since these sites were not found elsewhere in the BCJA1 genome, this also suggests that the major transcripts of this phage originate from the CDS 5–6 intergenic region (Fig. 2b).

a The sequence immediately downstream of the BCJA1c integrase gene showing potential sites involved in integration. b The intergenic region between repressor and antirepressor (cro) genes of BCJA1. Putative repressor-binding sites are boxed, while potential promoters are underlined. Note that there are four P Cro sequences and a single PRep
Transcription
The most obvious region for promoters is the intergenic region between the repressor and cro genes. Analysis of this region, using Softberry’s BPROM program and by visual screening, revealed sequences that exhibit significantly similarity to the consensus promoter TTGACA (N15-17) TATAAT (Fig. 2). The repressor gene contains a single potential promoter (P Rep ), while in the case of cro, four promoters have been tentatively identified. Two of the putative promoters (P Rep and PCro4) additionally exhibit the extended −10 region “TGN” (Burr et al. 2000; Mitchell et al. 2003; Fig. 2b). In addition to the bidirectional ρ-independent terminator mentioned above, another terminator (ΔG −6.9 kcal) is located between nucleotides 1129 and 1158, which presumably restricts transcriptional readthrough from gene 2 into the integrase. Three additional terminators were discovered elsewhere in the genome (Fig. 1).
Morphogenesis
Since the genome is terminally redundant, we assume that BCJA1 packages DNA by a head-full mechanism with the terminase complex initiating packaging at a pac site; unfortunately, the location of the latter is unknown. We propose, on the basis of sequence similarity, that the products of genes 33 and 34 encode the small and large subunits of the terminase complex, respectively. Interestingly, while the small subunit shows greatest similarity to prophage terminases in Bacillus and Clostridium species, the large subunit is most closely related to S. pneumoniae phage MM1 terminase. A high percentage of the genes involved in morphogenesis show homology to other phages facilitating their functional identification. CDSs 35–45 are involved in capsid morphogenesis, while CDSs 46–53 are associated with tail assembly. Polyacrylamide gel electrophoretic analysis of denatured BCJA1 virion proteins revealed at least ten structural proteins with masses ranging from 17–120 kDa (Jarrell et al. 1997). The two major proteins were 36.5 and 28 kDa and were predicted to be the major capsid and tail proteins, respectively. In silico analysis of CDS 40 reveals a 34.9-kDa protein with less than 40% sequence identity to the major capsid proteins of S. pneumoniae phage MM1 and Lactococcus lactis phage ul36 (Labrie and Moineau 2002). These results suggest that the BCJA1 capsid protein is not proteolytically modified at the time of or after prohead assembly.
The most likely major tail protein is encoded by CDS 46. Unfortunately, its molecular weight (16.9 kDa) is significantly less than the protein tentatively identified as the major tail protein on the basis of its molecular mass (gp 50, 28 kDa). On the basis of comparative mass, homology, and synteny, we propose that CDS 46 represents the major tail protein of BCJA1.
The product of CDS 53 most probably encodes a host-specificity protein or phage tail fiber. Interestingly, like the tail fiber proteins of the T7-like coliphages, the N-terminal region is much more conserved than the C-terminal ligand-binding domain (Kovalyova and Kropinski 2003). While the closest overall sequence similarity is to a Bacillus halodurans protein, iterative PSI-BLAST analysis revealed relationships in the carboxy region to large proteins in B. cereus phage phBC6A51 (NP_831679), Streptococcus agalactiae prophage λSa2 (NP_688832), Lactobacillus johnsonii prophage Lj965 (NP_958595, Ventura et al. 2004), and Streptococcus mitis phage SM1 (NP_862890, Ventura et al. 2004). In the latter, the similar sequence “pblB” was experimentally characterized as a platelet-binding protein (Bensing et al. 2001a, 2001b).
Discussion
While phages are the most abundant and probably the most diverse life forms on Earth (Rohwer 2003), relatively few viruses have been isolated and even fewer fully characterized against the extremophilic bacteria. This is not due to the lack of bacterial species growing in extreme environments. Quantitatively, the greatest numbers of sequenced tailed phages are for members of the bacterial phyla Proteobacteria and Firmicutes. What has emerged from phage genomic studies is the realization of the extent to which nonhomologous recombination has created genomic mosaics (Hendrix et al. 1999; Hendrix 2002) and has complicated our understanding of their taxonomy (Lawrence et al. 2002; Proux et al. 2002). This is also borne out from our analysis of BCJA1, which one might expect to be ecologically isolated and thus, less subject to horizontal gene transfer.
We initially predicted that most homologues of the BCJA1 proteins would occur among the five sequenced “Bacillus” genomes: B. anthracis (Read et al. 2003); B. cereus (Ivanova et al. 2003); B. halodurans (Takami et al. 2000); B. subtilis (Kunst et al. 1997) and Oceanobacillus iheyensis (Takami et al. 2002) or the fully characterized Bacillus phages, which include B. subtilis phages SPP1 (Alonso et al. 1997), PZA (Paces et al. 1989), B103 (Pecenkova et al. 1997), SPβc2 (Ravantti et al. 2004), and GA-1 (Salas 2004); and B. thuringiensis phage Bam35. This was not the case, though BCJA1 CDS 3–8 are collinear with B. halodurans prophage Bha35X/Bh1 genes BH3551–BH3546. The possible exception is the Cro homologue of BCJA1 (CDS 6), which does not display sequence homology to B. halodurans gene BH3548. Interestingly, the latter encodes a basic protein of 98 amino acids, containing a helix–turn–helix motif. This unannotated gene is spatially arranged relative to the putative Bha35X/Bh1 repressor gene in an identical orientation to that of BCJA1 CDS 6–7, suggesting that BH3548 probably encodes a Cro homologue. The other region displaying B. halodurans homologues is defined by BCJA1 CDS 50 (BH0961) CDS 53 (BH0962), and CDS 59 (BH0963). These genes in B. halodurans are found upstream of an amidase–holin pair, suggesting the presence of another defective prophage in the bacterial genome. This cluster of genes also occurs in O. iheyensis.
The high degree of similarity between Bacillus phage BCJA1c and Streptococcus phage and prophage genes was unexpected. Similarity is particularly evident (Table 1) in the genes involved in DNA replication, recombination, and morphogenesis. This is practically apparent using a dotplot comparison of the nucleotide sequence of BCJA1 with Streptococcus pyogenes prophage 370.1 (Ferretti et al. 2001; Fig. 1), which, in typical phage evolution format, shows blocks of homology separated by regions that do not share a common ancestor. Canchaya et al. (Canchaya et al. 2003) have proposed that prophage 370.1 is a member of the Sf11-like group of Siphoviridae employing pac-site DNA packaging. It has also been shown that the morphogenesis genes of prophage Bha35X/Bh1 are homologous to those of S. pyogenes prophage 315.5 (Banks et al. 2002;Beres et al. 2002). This adds further proof to the argument that horizontal gene transfer has occurred freely among the members of the bacterial phylum Firmicutes.
Proteins achieve conformational stability by means of covalent bonds (Cys-Cys), electrostatic forces, hydrogen bonds, and van der Waals interactions. These are all influenced by extremes of pH. We had hoped that a physicochemical comparison of alkaliphilic versus nonalkaliphilic viral structural homologues would provide us with some understanding of the nature of protein structure and its adaptation to high pH environments. Phage BCJA1 virions are completely stable from pH 6–11, but lose 75% of their titer after 1 h at pH 4 (Jarrell et al. 1997). Using the portal, major capsid, and major tail proteins as indicators, there was no significant difference in the relative concentrations of strongly acidic (D, E), strongly basic (K, R), hydrophobic (A, I, L, F, W, V) or polar amino acids (N, C, Q, S, T, Y) when comparing BCJA1 and its closest homologues. While within the window of pH stability, only the ionic interactions between charged side chains would be affected, other protein stabilizing interactions would function to achieve virion structural stability. These forces function in both alkaliphilic and nonalkaliphilic systems.
References
Abeles AL, Snyder KM, Chattoraj DK (1984) P1 plasmid replication: replicon structure. J Mol Biol 173:307–324
Agnew MD, Koval SF, Jarrell KF (1995) Isolation and characterization of novel alkaliphiles from bauxite-processing waste and description of Bacillus vedderi sp. nov., a new obligate alkaliphile. Syst Appl Microbiol 18:221–230
Alam SL, Atkins JF, Gesteland RF (1999) Programmed ribosomal frameshifting: much ado about knotting! Proc Natl Acad Sci USA 96:14177–14179
Alonso JC, Luder G, Stiege AC, Chai S, Weise F, Trautner TA (1997) The complete nucleotide sequence and functional organization of Bacillus subtilis bacteriophage SPP1. Gene 204:201–212
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–4022
Bankhead T, Segall AM (2000) Characterization of a mutation of bacteriophage λ integrase: putative role in core binding and strand exchange for a conserved residue. J Biol Sci 275:36949–36956
Banks DJ, Beres SB, Musser JM (2002) The fundamental contribution of phages to GAS evolution, genome diversification and strain emergence. Trends Microbiol 10:515–521
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL (2002) The Pfam protein families database. Nucleic Acids Res 30:276–280
Bensing BA, Rubens CE, Sullam PM (2001a) Genetic loci of Streptococcus mitis that mediate binding to human platelets. Infect Immun 69:1373–1380
Bensing BA, Siboo IR, Sullam PM (2001b) Proteins PblA and PblB of Streptococcus mitis, which promote binding to human platelets, are encoded within a lysogenic bacteriophage. Infect Immun 69:6186–6192
Beres SB, Sylva GL, Barbian KD, Lei B, Hoff JS, Mammarella ND, Liu MY, Smoot JC, Porcella SF, Parkins LD, Campbell DS, Smith TM, McCormick JK, Leung DY, Schlievert PM, Musser JM (2002) Genome sequence of a serotype M3 strain of group A Streptococcus: phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA 99:10078–10083
Betermier M, Galas DJ, Chandler M (1994) Interaction of Fis protein with DNA: bending and specificity of binding. Biochimie 76:958–967
Brendel V, Trifonov EN (1984) A computer algorithm for testing potential prokaryotic terminators. Nucleic Acids Res 12:4411–4427
Brendel V, Hamm GH, Trifonov EN (1986) Terminators of transcription with RNA polymerase from Escherichia coli: what they look like and how to find them. J Biomol Struct Dyn 3:705–723
Brüssow H, Bruttin A (1995) Characterization of a temperate Streptococcus thermophilus bacteriophage and its genetic relationship with lytic phages. Virology 212:632–640
Burr T, Mitchell J, Kolb A, Minchin S, Busby S (2000) DNA sequence elements located immediately upstream of the −10 hexamer in Escherichia coli promoters: a systematic study. Nucleic Acids Res 28:1864–1870
Campbell AM (1992) Chromosomal insertion sites for phages and plasmids. J Bacteriol 174:7495–7499
Canchaya C, Fournous G, Chibani-Chennoufi S, Dillmann ML, Brüssow H (2003) Phage as agents of lateral gene transfer. Curr Opin Microbiol 6:417–424
Chattoraj DK, Snyder KM, Abeles AL (1985) P1 plasmid replication: multiple functions of RepA protein at the origin. Proc Natl Acad Sci USA 82:2588–2592
Craig NL, Roberts JW (1980) E. coli recA protein-directed cleavage of phage lambda repressor requires polynucleotide. Nature 283:26–30
Esposito D, Scocca JJ (1997) The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res 25:3605–3614
Ferretti JJ, McShan WM, Ajdic D, Savic DJ, Savic G, Lyon K, Primeaux C, Sezate S, Suvorov AN, Kenton S, Lai HS, Lin SP, Qian Y, Jia HG, Najar FZ, Ren Q, Zhu H, Song L, White J, Yuan X, Clifton SW, Roe BA, McLaughlin R (2001) Complete genome sequence of an M1 strain of Streptococcus pyogenes. Proc Natl Acad Sci USA 98:4658–4663
Gertman E, White BN, Berry D, Kropinski AM (1986) IS222, a new insertion element associated with the genome of Pseudomonas aeruginosa. J Bacteriol 166:1134–1136
Goosen N, van de Putte P (1987) Regulation of transcription. In: Symonds N, Toussaint A, van de Putte P, Howe MM (eds) Phage Mu. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 41–52
Grigoriev A (1998) Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res 26:2286–2290
Grove A, Galeone A, Mayol L, Geiduschek EP (1996) Localized DNA flexibility contributes to target site selection by DNA-bending proteins. J Mol Biol 260:120–125
Grundling A, Bläsi U, Young R (2000) Biochemical and genetic evidence for three transmembrane domains in the class I holin, lambda S. J Biol Chem 275:769–776
Harger JW, Meskauskas A, Dinman JD (2002) An “integrated model” of programmed ribosomal frameshifting. Trends Biochem Sci 27:448–454
Hendrix RW (2002) Bacteriophages: evolution of the majority. Theor Popul Biol 61:471–480
Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF (1999) Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc Natl Acad Sci USA 96:2192–2197
Hofmann K, Bucher P, Falquet L, Bairoch A (1999) The PROSITE database, its status in 1999. Nucleic Acids Res 27:215–219
Horikoshi K, Yonezawa Y (1978) A bacteriophage active on an alkalophilic Bacillus sp. J Gen Virol 39:183–185
Ivanova N, Sorokin A, Anderson I, Galleron N, Candelon B, Kapatral V, Bhattacharyya A, Reznik G, Mikhailova N, Lapidus A, Chu L, Mazur M, Goltsman E, Larsen N, D’Souza M, Walunas T, Grechkin Y, Pusch G, Haselkorn R, Fonstein M, Ehrlich SD, Overbeek R, Kyrpides N (2003) Genome sequence of Bacillus cereus and comparative analysis with Bacillus anthracis. Nature 423:87–91
Jarrell KF, Vydykhan T, Lee P, Agnew MD, Thomas NA (1997) Isolation and characterization of bacteriophage BCJA1, a novel temperate bacteriophage active against the alkaliphilic bacterium, Bacillus clarkii. Extremophiles 1:199–206
Klein R, Baranyi U, Rossler N, Greineder B, Scholz H, Witte A (2002) Natrialba magadii virus phiCh1: first complete nucleotide sequence and functional organization of a virus infecting a haloalkaliphilic archaeon. Mol Microbiol 45:851–863
Kovalyova IV, Kropinski AM (2003) The complete genomic sequence of lytic bacteriophage gh-1 infecting Pseudomonas putida—evidence for close relationship to the T7 group. Virology 311:305–315
Kowalczuk M, Mackiewicz P, Mackiewicz D, Nowicka A, Dudkiewicz M, Dudek MR, Cebrat S (2001) DNA asymmetry and the replicational mutational pressure. J Appl Genet 42:553–577
Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Danchin A (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256
Labrie S, Moineau S (2002) Complete genomic sequence of bacteriophage ul36: demonstration of phage heterogeneity within the P335 quasi-species of lactococcal phages. Virology 296:308–320
Lavigne R, Sun WD, Volckaert G (2004) PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes. Bioinformatics 20:629–6135
Lawrence JG, Hatfull GF, Hendrix RW (2002) Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J Bacteriol 184:4891–4905
Letunic I, Goodstadt L, Dickens NJ, Doerks T, Schultz J, Mott R, Ciccarelli F, Copley RR, Ponting CP, Bork P (2002) Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res 30:242–244
Little JW (1991) Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73:411–421
Lobry JR (1999) Genomic landscapes. Microbiol Today 26:164–165
Mantri Y, Williams KP (2004a) Islander database. Available at http://129.79.232.60/cgi-bin/islander/islander.cgi
Mantri Y, Williams KP (2004b) Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic Acids Res 32 Database issue:D55–D58
Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, Liebert CA, Liu C, Madej T, Marchler GH, Mazumder R, Nikolskaya AN, Panchenko AR, Rao BS, Shoemaker BA, Simonyan V, Song JS, Thiessen PA, Vasudevan S, Wang Y, Yamashita RA, Yin JJ, Bryant SH (2003) CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res 31:383–387
Mitchell JE, Zheng D, Busby SJ, Minchin SD (2003) Identification and analysis of ‘extended −10’ promoters in Escherichia coli. Nucleic Acids Res 31:4689–4695
Nash HA (1990) Bending and supercoiling of DNA at the attachment site of bacteriophage λ. TIBS 15:222–227
Nielsen P, Fritze D, Priest FG (2004) Phenetic diversity of alkaliphilic Bacillus strains: proposal for nine new species. Microbiology 141:1745–1761
Nunes-Duby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res 26:391–406
Obregon V, Garcia JL, Garcia E, Lopez R, Garcia P (2003) Genome organization and molecular analysis of the temperate bacteriophage MM1 of Streptococcus pneumoniae. J Bacteriol 185:2362–2368
Ogawa T, Masukata H, Tomizawa J (1988) Transcriptional regulation of early functions of bacteriophage ϕ 80. J Mol Biol 202:551–563
Paces V, Vlcek C, Urbanek P, Hostomsky Z (1989) Nucleotide sequence of the right early region of Bacillus subtilis phage PZA completes the 19,366-bp sequence of PZA genome. Comparison with the homologous sequence of phage phi 29. Gene 44:115–120
Pande S, Vimaladithan A, Zhao H, Farabaugh PJ (1995) Pulling the ribosome out of frame by +1 at a programmed frameshift site by cognate binding of aminoacyl-tRNA. Mol Cell Biol 15:298–304
Pecenkova T, Benes V, Paces J, Vlcek C, Paces V (1997) Bacteriophage B103: complete DNA sequence of its genome and relationship to other Bacillus phages. Gene 199:157–163
Prangishvili D (2003) Evolutionary insights from studies on viruses of hyperthermophilic archaea. Res Microbiol 154:289–294
Proux C, van Sinderen D, Suarez J, Garcia P, Ladero V, Fitzgerald GF, Desiere F, Brussow H (2002) The dilemma of phage taxonomy illustrated by comparative genomics of Sfi21-like Siphoviridae in lactic acid bacteria. J Bacteriol 184:6026–6036
Rachel R, Bettstetter M, Hedlund BP, Haring M, Kessler A, Stetter KO, Prangishvili D (2002) Remarkable morphological diversity of viruses and virus-like particles in hot terrestrial environments. Arch Virol 147:2419–2429
Ravantti JJ, Duesterhoeft A, Soldo B, Hilbert H, Mauel C, Karamata D (2004) The complete nucleotide sequence of the Bacillus subtilis SPbetac2 prophage (GenBank accession number NC_001884).
Ravin NV, Kuprianov VV, Gilcrease EB, Casjens SR (2003) Bidirectional replication from an internal ori site of the linear N15 plasmid prophage. Nucleic Acids Res 31:6552–6560
Raymond-Denise A, Guillen N (1991) Identification of dinR, a DNA damage-inducible regulator gene of Bacillus subtilis. J Bacteriol 173:7084–7091
Read TD, Peterson SN, Tourasse N, Baillie LW, Paulsen IT, Nelson KE, Tettelin H, Fouts DE, Eisen JA, Gill SR, Holtzapple EK, Okstad OA, Helgason E, Rilstone J, Wu M, Kolonay JF, Beanan MJ, Dodson RJ, Brinkac LM, Gwinn M, DeBoy RT, Madpu R, Daugherty SC, Durkin AS, Haft DH, Nelson WC, Peterson JD, Pop M, Khouri HM, Radune D, Benton JL, Mahamoud Y, Jiang L, Hance IR, Weidman JF, Berry KJ, Plaut RD, Wolf AM, Watkins KL, Nierman WC, Hazen A, Cline R, Redmond C, Thwaite JE, White O, Salzberg SL, Thomason B, Friedlander AM, Koehler TM, Hanna PC, Kolsto AB, Fraser CM (2003) The genome sequence of Bacillus anthracis Ames and comparison to closely related bacteria. Nature 423:81–86
Reeder J, Giegerich R (2004) Design implementation and evaluation of a practical pseudoknots folding algorithm based upon thermodynamics. BMC Bioinform 5:104–115
Roberts JW, Roberts CW, Mount DW (1977) Inactivation and proteolytic cleavage of phage lambda repressor in vitro in an ATP-dependent reaction. Proc Natl Acad Sci USA 74:2283–2287
Rohwer F (2003) Global phage diversity. Cell 113:141
Salas M (2004) Bacillus phage GA-1 virion, complete genome (GenBank accession number NC_002649)
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Schultz J, Copley RR, Doerks T, Ponting CP, Bork P (2000) SMART: a web-based tool for the study of genetically mobile domains. Nucleic Acids Res 28:231–234
Shine J, Dalgarno L (1974) The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci USA 71:1342–1346
Shine J, Dalgarno L (1975) Terminal-sequence analysis of bacterial ribosomal RNA. Correlation between the 3′-terminal-polypyrimidine sequence of 16-S RNA and translational specificity of the ribosome. Eur J Biochem 57:221–230
Slepecky RA, Hemphill RA (1992) The genus Bacillus—nonmedical. In: Balows A, Truper HG, Dworkin M, Harder W, Schleifer K-H (eds) The Prokaryotes. A handbook on the biology and bacteria: ecophysiology, isolation, identification, applications. Springer, Berlin Heidelberg New York, pp 1663–1696
Sonnhammer ELL, von Heijne G, Krogh A (1998) A hidden Markov model for predicting transmembrane helices in protein sequences. In: Glasgow J, Littlejohn T, Major F, Lathrop R, Sankoff D, Sensen C (eds) Proceedings of the 6th international conference on intelligent systems for molecular biology. AAAI, Menlo Park, pp 175–182
Stanley E, Fitzgerald GF, Le Marrec C, Fayard B, van Sinderen D (1997) Sequence analysis and characterization of phi O1205, a temperate bacteriophage infecting Streptococcus thermophilus CNRZ1205. Microbiology 143:3417–3429
Studholme DJ, Dixon R (2003) Domain architectures of sigma54-dependent transcriptional activators. J Bacteriol 185:1757–1767
Swinger KK, Lemberg KM, Zhang Y, Rice PA (2003) Flexible DNA bending in HU-DNA cocrystal structures. EMBO J 22:3749–3760
Takami H, Nakasone K, Takaki Y, Maeno G, Sasaki R, Masui N, Fuji F, Hirama C, Nakamura Y, Ogasawara N, Kuhara S, Horikoshi K (2000) Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res 28:4317–4331
Takami H, Takaki Y, Uchiyama I (2002) Genome sequence of Oceanobacillus iheyensis isolated from the Iheya Ridge and its unexpected adaptive capabilities to extreme environments. Nucleic Acids Res 30:3927–3935
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA (2003) The COG database: an updated version includes eukaryotes. BMC Bioinform 4:41
Unniraman S, Prakash R, Nagaraja V (2002) Conserved economics of transcription termination in eubacteria. Nucleic Acids Res 30:675–684
Ventura M, Canchaya C, Pridmore RD, Brüssow H (2004) The prophages of Lactobacillus johnsonii NCC 533: comparative genomics and transcription analysis. Virology 320:229–242
Walker JE, Saraste M, Runswick MJ, Gay NJ (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951
Young R (1992) Bacteriophage lysis: mechanism and regulation. Microbiol Rev 56:430–481
Young R, Bläsi U (1995) Holins: form and function in bacteriophage lysis. FEMS Microbiol Rev 17:191–205
Acknowledgements
A.K. and K.J. acknowledge research funding from the Natural Sciences and Engineering Research Council of Canada.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Horikoshi
Rights and permissions
About this article
Cite this article
Kropinski, A.M., Hayward, M., Agnew, M.D. et al. The genome of BCJA1c: a bacteriophage active against the alkaliphilic bacterium, Bacillus clarkii. Extremophiles 9, 99–109 (2005). https://doi.org/10.1007/s00792-004-0425-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00792-004-0425-0