
Abstract
Bone is a connective tissue and guarantees protection and support of organ function. Contrary to the common view, bone is a dynamic tissue that constantly undergoes turnover in order to maintain stability and integrity. In this process called bone turnover or bone remodelling, two effector cell types are involved. Osteoclasts, specialised for bone resorption, and osteoblasts, responsible for bone formation, are key players in bone turnover. In the past decade, a lot of information about signal pathways, osteoblast–osteoclast communication and osteoclast activation concerning bone remodelling has arisen. In this publication, we aim to review molecular and biochemical insights with respect to the bone remodelling process. The bone remodelling process is of fundamental importance for craniofacial growth, orthodontic tooth movement and regenerative dentistry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
General function and structure of bone
Bone is a hallmark of all vertebrates and absolutely essential in terms of organ protection and support, brain and lung function, locomotion, support of haematopoiesis in the bone marrow, storage of minerals (i.e. calcium, phosphate) and providing attachment to muscles. Bone tissue is largely composed of type I collagen amounting to about 90% of total bone protein. The remaining organic component consists of non-structural proteins like growth factors, blood protein, osteonectin and osteocalcin. The inorganic phase of bone is mainly composed of mineral hydroxyappatite (Ca10(PO4)6(OH)2). Due to its material consistency, bone has a relatively flexible character as well as compressive strength. The mature bone is composed of two different types of tissue: The hard outer layer of bone, the so-called cortical bone, is responsible for the stability of the skeleton. This tissue appears smooth, white and solid. The cortical bone contains osteons (Haversian systems), which are composed of a central canal (Haversian canal) surrounded by lamellae of bone matrix. Within the lamellae, there are osteocytes embedded in tiny spaces (lacunae). The Haversian canal encompasses blood vessels and nerve cells throughout the bone and communicates with osteocytes in lacunae through canaliculi. Moreover, the corticalis has an outer membrane, the periosteum. The periosteum consists of an outer fibrous layer and an inner one that has osteogenic potential and enables the bone to enlarge [31]. The inside of bone is assembled by a trabecular network (spongiosa) and harbours bone marrow or embryonic connective tissue. The spongiosa ensures elasticity and stability of the skeleton and accounts for the main part (about 70%) of bone metabolism [50].
Bone remodelling
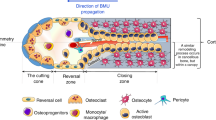
In order to maintain stability and integrity of bone, it is constantly undergoing remodelling, with about 10% of bone material being renewed each year [40]. Bone remodelling is a complex process that involves bone resorption performed by osteoclasts, followed by bone formation carried out by osteoblasts. In this process, these cells closely collaborate in basic multicellular units (BMU) [23, 25, 50]. The bone remodelling cycle involves different sequential steps, namely resorption, reversal and formation (Fig. 1) [23, 25]. The osteoclast stimulation occurs in BMU, which are small cortical and trabecular bone covering areas, where the remodelling process takes place. In these sites, osteoclastogenesis precedes with the expression of receptor activator of nuclear factor κ B ligand (RANKL) by bone lining cells, which in turn binds to the RANK that exists as a surface receptor on the membrane of pre-osteoclasts. This binding promotes an intricate and distinct signalling cascade for osteoclast activation and commitment [70]. The expression of RANKL is up-regulated in the presence of interleukin-1 (IL-1), tumour necrosis factor α (TNF-α) or vitamin D, whereas transforming growth factor ß (TGFß) or estrogens have an opposite effect. It is recognised that the expression of RANKL is up-regulated in malignant tumour cells, where it is involved in bone destruction. In the resorption stage, chemokines or chemotactic cytokines (MCP-1 or monocyte chemoattractant protein-1) are secreted by stromal cells or bone lining cells to attract and stimulate recruitment of osteoclast precursors for bone resorption. Furthermore, MCP-1 is produced in response to the presence of parathyroid hormone or inflammatory cytokines such as TNF-α and Interleukin-1ß [22, 41, 46]. Regarding bone turnover, osteocytes are considered to influence bone remodelling. Osteocytes, in bone embedded spider-shaped cells, have been proposed to sense microcracks and microfractures, which trigger subsequent osteoclast differentiation and bone resorption [6, 46]. Osteocytes are connected with other cells on the bone surface or to each other by cytoplasmic processes, which traverse the bone. It is thought that osteocytes can regulate the expression of RANKL in osteoblasts, thus, influencing osteoclastogenesis [7]. It was demonstrated that radio-labelled parathyroid hormone localises to osteocytes, where PTH suppresses the expression of sclerostin, which is exclusively synthesised by osteocytes. Sclerostin exerts an antagonising action on the wnt signalling pathway by binding to the LRP5/6 receptor, which results in an inhibition of osteoblastogenesis and in an increased osteoblast apoptosis. Thus, osteocytes exert a negative regulation of osteoblast number and formation of bone by secreting sclerostin [19]. A special receptor for PTH, parathyroid hormone receptor 1 (PTHR1), is expressed in osteocytes and in osteoblasts [16]. O’Brien et al. [51] demonstrated that the activation of PTHR1 in transgenic mice decreases the expression of sclerostin, which results in a higher bone mass. However, a directed activation of the osteocyte specific PTHR1 was unexpectedly accompanied by an increase of osteoclast number and accelerated bone resorption, which was also manifested by an elevated level of RANKL and M-CSF. Nevertheless, the PTHR1 activation in osteocytes led to a higher rate of bone remodelling with a positive balance resulting in more bone [51]. In the further course of resorption, the osteoclast progenitors are differentiated to fully active osteoclasts that dig trenches with a depth of 40–60 µm in the bone [23]. The possibility has been discussed that during bone resorption liberated matrix, proteins from the bone may involved in osteoblast commitment, such as bone morphogenetic proteins (BMPs) and insuline-like growth factors -II, which in turn would reverse the process from bone resorption to bone formation. However, this assumption is questionable because of the matrix protein degrading effect of cathepsin-K and other proteases, which are secreted by the osteoclasts. In addition, some evidences were found that the coupling of liberated growth factors from the bone is dispensable for osteoblast commitment [46]. The resorption phase is followed by the reversal phase comprising the differentiation of osteoblast precursors and the discontinuation of bone resorption with osteoclast apoptosis. Osteoblasts become probably activated by secretion products from osteoclasts [46]. The osteoblast activating effect of osteoclast secretion products like sphingosine 1-phosphate (S1P), myb-induced myeloid protein-1 (mim-1), a B polypeptide chain platelet-derived growth factor homodimer (PDGF-BB) and a hepatocyte growth factor (HGF) has been demonstrated [46]. Secreted S1P binds to an osteoblast S1P receptor, which results to an up-regulation of RANKL expression and in an enhanced osteoblast survival [46]. PDGF-BB supports a better proliferation of osteoblast pre-cursors, whereas the differentiation of these pre-cursors is suppressed. Secreted HGF from osteoclasts stimulates DNA synthesis and proliferation in osteoblasts as well in osteoclasts by binding to HGF receptor that is found in both cell types [46]. After osteoblast activation, osteoblasts lay down new bone material (i.e. collagen type I, osteocalcin, osteopontin, etc.) until the resorbed bone is entirely replaced by a new one [25]. Osteoblasts that become encased in the new bone matrix during bone formation are transformed to osteocytes, the most abundant cellular component of the bone [17]. The osteoblasts become quiescent at the end of bone remodelling, and they form flattened lining cell on the bone surface until a new remodelling cycle is triggered. The osteocyte product sclerostin may involve in this process by antagonising the canonical wnt signalling pathway, which leads to a reduced osteoblast activity. Osteopetrotic diseases like Van Buchem disease and sclerosteosis are caused by a loss of function mutation of the sost gene product sclerostin. Therefore, sclerostin is regarded as a key inhibitor that regulates the normal extent of bone formation and consequently protects against the deleterious effects of uncontrolled bone formation [55].

Overview of bone remodelling. Resting phase—the bone surface is coated by resting flattened lining stromal cells. Resorption phase—after triggering the remodelling process, osteoclasts degrade old bone matrix. Reversal phase and bone formation—after bone removal, osteoclasts undergo apoptosis and osteoblasts become activated and lay down new bone material in the trench. Resting phase—with the replacement of old bone by new one, the osteoblasts form resting flattened lining cells on the surface of bone
Osteoblasts: regulation and bone formation
Osteoblasts are effector cells for bone formation with the widely known ability to form bone tissue by secretion of alkaline phosphatase, type I collagen, proteoglycan, bone sialoprotein and osteopontin. Osteoblasts do not function individually but are found in clusters along the bone surface where they produce bone matrix by deposition of collagen 1. Subsequently, the collagen matrix becomes mineralised, presumably through the activity of alkaline phosphatase. Although the precise function of alkaline phosphatase is less clear, it has been assumed to be involved in the bone mineralisation process [71]. Besides their role in bone formation, osteoblasts are also involved in osteoclast differentiation as they produce RANKL or OPG to modulate osteoclast activity [62]. Osteoblast cells originate from pluripotent mesenchymal stem cells of the bone marrow [31]. A number of transcription factors and biochemical factors tightly regulate osteoblast recruitment, function and maturation. Osteoblast differentiation is promoted by the secretion of lipid-modified glycoproteins of the wingless (wnt) family, BMP-2 and several transcription factors.
In order to give a general survey, we aim to describe important signalling pathways that induce osteoblastogenesis and to define essential transcription factors such as Runx2 and Osterix.
Canonical wnt pathway
Wnt proteins are involved in signalling pathways during development and later stages of life. Regarding bone formation, wnt proteins activate a specific type of the intracellular signalling cascade called the canonical wnt signalling or wnt/ß-catenin pathway (Fig. 2) [54]. Binding of a secreted wnt protein through a low-density lipoprotein receptor-related protein 5 and 6 (LRP5/6) co-receptor leads to the disruption of a protein complex, consisting of adenomatous polyposis coli (APC), axin and glycogen synthase kinase 3 (GSK3). Consequently, the phosphorylation of ß-catenin by GSK3 is inhibited, thus, causing hypophosphorylation of ß-catenin. In this way, stabilised ß-catenin is accumulated in the cytoplasm and translocated to the nucleus where it interacts with T cell factor (TCF)/lymphoid enhancer binding factor transcription factors to support gene expression of wnt-responsive genes [5, 37]. In the absence of wnt, APC and axin act as a scaffold protein complex to support the phosphorylation activity and binding of GSK3 to the substrate ß-catenin, whereas phosphorylated ß-catenin is marked for degradation by the ubiquitin-dependent proteasome [37]. In fact, suppression of GSK3 activity by an artificial inhibitor (e.g. CHIR99021) caused an increase of bone mass [4]. In another case, an inactivating mutation of the wnt-specific co-receptor LRP5/6 caused osteoporosis [21]. Furthermore, results from quantitative PCR analysis pointed out that elevated wnt10b concentrations in a bone-marrow-derived cell culture was proportional to an increase of osteoblastogenic transcription factors (e.g. Osterix, Runx2) [4]. Thus, it has become clear that the wnt protein-induced signal cascade stimulates osteoblastogenesis. A number of secreted antagonist proteins regulate the canonical wnt pathway: For example, the antagonists of wnt protein are the secreted frizzled-related proteins, wnt inhibitory factor-1 that binds to wnt protein and prevents wnt activity [73] and members of the dickkopf family [45]. Dickkopf binds to LRP5/6 to make up a complex with the transmembrane Kremens protein (kringle-containing protein marking the eye and the nose) that trigger the removal of LRP5/6 from the cell surface [37, 45]. Thus, the number of wnt-specific receptors is reduced. Sclerostin (a SOST gene product), another antagonist of wnt protein and negative regulator of bone formation, binds to the LRP5/6 receptor and prevents binding of wnt protein [37, 68].

Overview over the canonical wnt pathway. In the absence of wnt, the scaffold protein complex promotes the hyperphosphorylation of ß-catenin by GSK, whereby ß-catenin becomes degraded by the ubiquitin-dependent proteasome. The binding of wnt to the frizzled receptor results in activation of dishevelled and to an interruption of the scaffold proteins, axin and APC. This results in a hypophosphorylation of ß-catenin, which allows translocation of ß-catenin into the nucleus where it becomes associated with the TCF-transcription factor and participates in the initiation of transcription of target genes
Bone morphogenetic proteins
BMP belongs to the large super family of TGFß. BMPs were extensively researched and were demonstrated to play an important role during embryonic development [30] as well as in cartilage and bone formation [8]. For instance, some BMPs were found in bone [67]. In vitro studies involving cell cultures have shown that BMPs are able to convert the differentiation pathway of myoblastic cell lines into that of osteoblast lineage. Furthermore, it was demonstrated that the expression of osteoblast specific proteins, such as alkaline phosphatase osteocalcin and the osteogenic transcription factor Runx2, is induced by BMPs [31, 32]. BMPs induce osteoblast differentiation through a Smad dependent signal pathway (Fig. 3): The secreted BMP dimer binds cooperatively to types I and II serine/threonine kinase receptor. Upon binding of BMPs to type II receptor, type I receptor kinase is phosphorylated by the activated type II receptor kinase. Receptor-regulated Smad (R-Smad; Smad 1, 2, 3, 5 and 8) proteins interact transiently with type I receptor kinase and become phosphorylated at SSXS motif at their C-termini [36]. Consequently, these phosphorylated R-Smad proteins associate with the common partner co-Smad protein (Smad 4) to form a heteromeric complex that is subsequently translocated into the nucleus. There, they interact with transcription factors, for example with Runx2, to participate in gene transcription [53]. Additionally, it has been reported that an inhibitory class of Smad proteins, i.e. Smad 6 and 7, interacts with phosphorylated type kinase I or with co-Smad and, thus, prevents the activation of R-Smad or reduces the scale of R-Smad/co-Smad complex formation [2].

Overview of the TGFß superfamily signalling pathway. Upon BMP activation, type II receptors transphosphorylate type I receptors. Type I receptor phosphorylates R-Smad proteins, which in turn associate with Smad-4 (co-Smad) in a complex. This complex moves across the nuclear membrane into the nucleus, where the Smad–protein complex induces specific gene expression
Transcription factors that regulate osteoblast differentiation
Runx2/cfba1, a member of the Runt-related family of transcription factors, is an important transcription factor for osteoblast differentiation. As mentioned, signal pathways like wnt/ß-catenine and the TGF-ß/BMP pathway can modulate the transcriptional factor to induce an osteogenic phenotype [10, 18, 27]. It has been shown that null mutants of mice that do not express Runx2 fail to form any bone tissue and that osteoblast maturation is blocked [34]. Moreover, Ducy [13] demonstrated that Runx2 can activate osteoblast-specific genes in non-osteoblastic cells and that the expression rates of osteocalcin, osteopontin, alkaline phosphatase and type I collagen genes were variably expressed when cells were transfected with different isoforms of Runx2 [24]. Thus, it is obvious that transcription factor Runx2 plays a significant role for osteoblastogenesis. Osterix, a zinc finger-containing transcription factor, is similar to Runx2, an essential transcription factor for osteoblast differentiation. Studies made by Nakashima et al. [48] have demonstrated that Osterix-null mutants of mice show a similar phenotype like Runx2-null mutants in terms of the lack of bone formation owing to the absence of osteoblast differentiation. It was noted that in osteogenic cells of Osterix-null mutant, the transcription level of Runx2 was in the same scale with those of wild-type osteoblasts [48]. However, no Osterix transcripts were detected in Runx2-null mice [49]. Therefore, it is assumed that Osterix must be acting downstream of Runx2 in the differentiation pathway of osteoblasts.
Osteoclasts: regulation and bone resorption
Osteoclasts are multi-nucleated cells specialised in bone resorption. The first step is involved in recruitment and dissemination of osteoclast progenitors, stemming from haematopoietic monocyte-macrophage lineage [38, 57]. It was noted that osteoclast activity and formation is dependent from the presence of two different cytokines, macrophage colony-stimulating factors (M-CSF) and RANKL and calciotropic hormones (Fig. 4) [31, 64]. M-CSF binds directly to its receptor, c-fms, on the surface of the early osteoclast progenitor and, thereby, provides signals required for proliferation [64]. Furthermore, M-CSF enhances osteoclast activity owing to prevent osteoclast apoptosis [20, 57]. Indeed, Yoshida [74] demonstrated the important role of M-CSF in rodents, where mutations in the coding region of M-CSF caused a severe osteopetrotic phenotype due to the functionally inactivated M-CSF. However, administration of M-CSF to the rodents with inactivated M-CFS led to the restoration of impaired bone resorption [15, 28, 33]. As mentioned, the second important osteoclast key activator is RANKL. RANKL is a member of the TNF gene family, which is predominantly expressed on the surface of osteoblast stromal cells. Systemic and local factors like parathyroid hormone, IL-1 or prostaglandins can induce osteoclast formation by increasing expression of RANKL on the surface of immature osteoblasts and marrow stromal cells [10, 31, 35, 39, 57, 61]. The osteoclast differentiation by RANKL and its RANK is accomplished by cell–cell interaction between osteoblasts and osteoclasts. RANKL that is membrane bound on the osteoblast surface thereby binds to RANK, which is placed on the surface of the osteoclast [56, 65]. For instance, it has been observed for decades that in vitro differentiation of monocytes to osteoclasts occurs only with co-cultured osteoblasts [63]. Moreover, studies by Odgren [52] and Dougall [12] pointed out the crucial role of RANK and RANKL concerning osteoclast activation. Thus, cell-to-cell signalling by RANKL and RANK is necessary for osteoclast differentiation. Another cytokine that is expressed by osteoblasts, osteoprotegrin (OPG) acts as decoy receptor for RANKL and, thereby, inhibits osteoclast differentiation [60, 72]. Thus, it was assumed that the proportion between RANKL and OPG could regulate bone resorption and bone strength [35]. Under inflammatory conditions, such as rheumatoid arthritis or myeloma, the proportion between RANKL to OPG is increased, thus, favouring increased osteoclastogenesis [57]. Three calciotropic hormones acting in different ways control osteoclast activity [56]: Vitamin D promotes the differentiation of osteoclasts from monocyte macrophage stem cell precursors in vitro. In vivo, enhanced osteoclastic bone resorption was observed when vitamin D was applied at high doses [26, 66]. This effect is caused by the stimulation of RANKL production by osteoblasts. Parathyroid hormone is a peptide secreted by cells of the parathyroid gland [56]. It is secreted in response to changes in blood calcium and affects bone formation and resorption. It acts antagonistically to calcitonin by increasing the concentration of calcium in blood plasma. Parathyroid hormone binds to osteoblasts and induces the production of surface proteins (M-CSF and RANKL) that stimulate the maturation and action of osteoclasts [56]. Calcitonin is synthesised primarily by the parafollicular C-cells of the thyroid in response to the calcium concentration within the C-cells. Several studies concluded that calcitonin inhibits bone resorption by acting directly on osteoclasts through its receptor [9, 56].

Overview of cell–cell interaction between osteoblast and osteoclast. M-CSF acts on osteoclast precursors to control their proliferation and differentiation. The osteoblasts produce a protein called receptor activator of nuclear factor kappa B ligand; this can bind to the receptor RANK that is localised on the osteoclast precursor and stimulate it to differentiate to fully activated osteoclast. OPG that is secreted by osteoblasts competes with RANK for RANKL. Parathyroid hormone (PTH), interleukin-1 (IL-1), prostaglandin (PGE2) and 1,25 (OH) 2 vitamin D 3 are stimulators of bone resorption in that they stimulate the expression of RANKL in osteoblast cells
The initial step of bone resorption is migration of osteoclasts to the bone area to be remodelled, where they become adherent to the bone. The attachment of osteoclasts is accomplished by formation of podosomes containing filamentous actin and αvβ3 integrin, which interact with bone matrix proteins, such as osteopontin and vitronectin [14, 47, 66]. The intimate contact between the bone matrix and the osteoclasts is reflected by the polarisation of the latter, with the plasma membrane forming three distinct areas: first, the basolateral membrane which is not in contact with bone material; second, the sealing zone that is closely attached to the bone; and third, the ruffled border which is ring-shaped and surrounded by the sealing zone. The ruffled border faces the bone matrix and is in direct contact with the matrix. The sealing zone forms a diffusion barrier to ensure a defined proton and protease concentration secreted by the ruffled border in the evolving resorption lacunae underneath the osteoclast cell [69]. Active osteoclasts form a resorptive front side, attaching to the bone matrix. Osteoclasts are capable of dissolving bone material, such as mineral hydroxyapatite, by secretion of hydrochloric acid. A V-type ATPase, which is present in the ruffled membrane of osteoclasts, translocates protons into the resorption lacuna and, thus, acidifies the environment (pH ∼ 4.5). Moreover, a carbonic anhydrase II is present in osteoclasts to supply the need of protons. To ensure electro neutrality, a chloride channel (CLC7) simultaneously transports the counter ion chloride [69]. Because of demineralisation of bone material, the organic phase of bone becomes more accessible. It is assumed that lyosomal cathepsin K and, to a lesser extent, matrix metalloproteinases are responsible for the degradation of organic bone matrix. Present data show that cathepsin K plays a major role in degradation of type-1 collagen under acidic conditions [58]. In contrast, matrix metallo proteinase-9 (gelatinase B or MMP9), a second abundant protease, from osteoclastic origin barely degrade the organic matrix. Rather, MMP-9 is considered to be involved in the initiation of bone degradation by the removal of the collagenous layer from the bone surface and cleaning the resorption pits from remaining collagen after cathepsin K digestion [11, 42]. During bone resorption, a high concentration of calcium up to 40 mM was observed in the microenvironment of the resorption site [59] that inhibits osteoclast activity [43]. The rise of calcium results in an alteration of the calcium level in the osteoclasts is followed by actin reorganisation and podosomal disassembly, and, consequently, leads to apoptosis [43, 44]. Even TGF-ß that blocks bone resorption has been reported to promote osteoclast apoptosis, which is reflected in nuclear and cytoplasmic condensation and fragmentation of DNA [25]. Besides calcium and TGF-ß, other substances like estrogens, some kinds of bisphophanats and taxomifen were able to induce osteoclast apoptosis [1, 3, 29].
Conclusion
Bone is a connective tissue designed to withstand mechanical forces. It undergoes remodelling in certain time intervals or in response to microdamages. Bone turnover is a complex process influenced by the mutual actions of osteoblasts and osteoclasts, which effect bone renewal. A variety of factors such as hormones and cytokines is involved in the differentiation and regulation of osteoblast and osteoclast cells. Disturbances in signal pathways, bone responsive transcription factors that affect bone remodelling can cause severe bone diseases (e.g. osteopetrosis, osteoporosis). The understanding of the molecular and biochemical mechanisms that enable the bone to maintain stability and integrity is essential to devising therapeutic strategies in all fields of regenerative medicine and dentistry.
References
Arnett TR, Lindsay R, Kilb JM, Moonga BS, Spowage M, Dempster DW (1996) Selective toxic effects of tamoxifen on osteoclasts: comparison with the effects of oestrogen. J Endocrinol 149:503–508
Balemans W, Van Hul W (2002) Extracellular regulation of BMP signaling in vertebrates: a cocktail of modulators. Review. Dev Biol 250:231–250
Benford HL, McGowan NW, Helfrich MH, Nuttall ME, Rogers MJ (2001) Visualization of bisphosphonate-induced caspase-3 activity in apoptotic osteoclasts in vitro. Bone 28:465–473
Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA (2005) Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci USA 102:3324–3329
Bodine PV, Komm BS (2006) Wnt signaling and osteoblastogenesis. Review. Rev Endocr Metab Disord 7:33–39
Bonewald LF (2007) Osteocytes as dynamic multifunctional cells. Review. Ann N Y Acad Sci 1116:281–290
Boyce BF, Yao Z, Zhang Q, Guo R, Lu Y, Schwarz EM, Xing L (2007) New roles for osteoclasts in bone. Review. Ann N Y Acad Sci 1116:245–254
Cao X, Chen D (2005) The BMP signaling and in vivo bone formation. Gene 357:1–8
Chambers TJ, Moore A (1983) The sensitivity of isolated osteoclasts to morphological transformation by calcitonin. J Clin Endocrinol Metab 57:819–824
Datta HK, Ng WF, Walker JA, Tuck SP, Varanasi SS (2008) The cell biology of bone metabolism. J Clin Pathol 61:577–587
Delaissé JM, Engsig MT, Everts V, del Carmen Ovejero M, Ferreras M, Lund L, Vu TH, Werb Z, Winding B, Lochter A, Karsdal MA, Troen T, Kirkegaard T, Lenhard T, Heegaard AM, Neff L, Baron R, Foged NT (2000) Proteinases in bone resorption: obvious and less obvious roles. Clin Chim Acta 291:223–234
Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, Armstrong A, Shen V, Bain S, Cosman D, Anderson D, Morrissey PJ, Peschon JJ, Schuh J (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Duong LT, Lakkakorpi P, Nakamura I, Rodan GA (2000) Integrins and signaling in osteoclast function. Matrix Biol 19:97–105
Felix R, Cecchini MG, Fleisch H (1990) Macrophage colony stimulating factor restores in vivo bone resorption in the op/op osteopetrotic mouse. Endocrinology 127:2592–2604
Fermor B, Skerry TM (1995) PTH/PTHrP receptor expression on osteoblasts and osteocytes but not resorbing bone surfaces in growing rats. J Bone Miner Res 10:1935–1943
Franz-Odendaal TA, Hall BK, Witten PE (2006) Buried alive: how osteoblasts become osteocytes. Dev Dyn 235:176–190
Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, Javed A, van Wijnen AJ, Stein JL, Stein GS, Lian JB (2005) Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem 280:33132–33140
Glass DA 2nd, Karsenty G (2007) In vivo analysis of Wnt signaling in bone. Endocrinology 148:2630–2634
Glantschnig H, Fisher JE, Wesolowski G, Rodan GA, Reszka AA (2003) M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ 10:1165–1177
Gong Y, Slee RB, Fukai N et al (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 107:513–523
Graves DT, Alsulaimani F, Ding Y, Marks SC Jr (2002) Developmentally regulated monocyte recruitment and bone resorption are modulated by functional deletion of the monocytic chemoattractant protein-1 gene. Bone 31:282–287
Hadjidakis DJ, Androulakis II (2006) Bone remodeling. Ann N Y Acad Sci 1092:385–396
Harada H, Tagashira S, Fujiwara M, Ogawa S, Katsumata T, Yamaguchi A, Komori T, Nakatsuka M (1999) Cbfa1 isoforms exert functional differences in osteoblast differentiation. J Biol Chem 274:6972–6978
Hill PA (1998) Bone remodelling. Br J Orthod 25:101–107
Hofbauer LC, Heufelder AE (2001) Role of receptor activator of nuclear factor-kappaB ligand and osteoprotegerin in bone cell biology. J Mol Med 79:243–253
Hogan BL (1996) Bone morphogenetic proteins in development. Curr Opin Genet Dev 6:432–438
Horwood NJ, Elliott J, Martin TJ, Gillespie MT (1998) Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology 139:4743–4766
Kameda T, Mano H, Yuasa T, Mori Y, Miyazawa K, Shiokawa M, Nakamaru Y, Hiroi E, Hiura K, Kameda A, Yang NN, Hakeda Y, Kumegawa M (1997) Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J Exp Med 186:489–495
Kassem M, Abdallah BM, Saeed H (2008) Osteoblastic cells: differentiation and trans-differentiation. Arch Biochem Biophys 473:183–187
Katagiri T, Takahashi N (2002) Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis 8:147–159
Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawa-Sehara A, Suda T (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol 127:1755–1766
Kodama H, Yamasaki A, Nose M, Niida S, Ohgame Y, Abe M, Kumegawa M, Suda T (1991) Congenital osteoclast deficiency in osteopetrotic (op/op) mice is cured by injections of macrophage colony-stimulating factor. J Exp Med 173:269–272
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M et al (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89:755–764
Kostenuik PJ (2005) Osteoprotegerin and RANKL regulate bone resorption, density, geometry and strength. Curr Opin Pharmacol 5:618–625
Kretzschmar M, Liu F, Hata A, Doody J, Massagué J (1997) The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev 11:984–995
Krishnan V, Bryant HU, Macdougald OA (2006) Regulation of bone mass by Wnt signaling. J Clin Invest 116:1202–1209
Kurihara N, Suda T, Miura Y, Nakauchi H, Kodama H, Hiura K, Hakeda Y, Kumegawa M (1989) Generation of osteoclasts from isolated hematopoietic progenitor cells. Blood 74:1295–1302
Lee SK, Lorenzo JA (1999) Parathyroid hormone stimulates TRANCE and inhibits osteoprotegerin messenger ribonucleic acid expression in murine bone marrow cultures: correlation with osteoclast-like cell formation. Endocrinology 140:3552–3561
Lerner UH (2006) Bone remodeling in post-menopausal osteoporosis. J Dent Res 85:584–595
Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC (2007) Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. J Biol Chem 282:33098–33106
Logar DB, Komadina R, Prezelj J, Ostanek B, Trost Z, Marc J (2007) Expression of bone resorption genes in osteoarthritis and in osteoporosis. J Bone Miner Metab 25:219–225
Lorget F, Kamel S, Mentaverri R, Wattel A, Naassila M, Maamer M, Brazier M (2000) High extracellular calcium concentrations directly stimulate osteoclast apoptosis. Biochem Biophys Res Commun 268:899–903
Malgaroli A, Meldolesi J, Zallone AZ, Teti A (1989) Control of cytosolic free calcium in rat and chicken osteoclasts. The role of extracellular calcium and calcitonin. J Biol Chem 264:14342–14347
Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C (2002) Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 417:664–667
Matsuo K, Irie N (2008) Osteoclast–osteoblast communication. Arch Biochem Biophys 473:201–209
Miyauchi A, Alvarez J, Greenfield EM, Teti A, Grano M, Colucci S, Zambonin-Zallone A, Ross FP, Teitelbaum SL, Cheresh D et al (1991) Recognition of osteopontin and related peptides by an alpha v beta 3 integrin stimulates immediate cell signals in osteoclasts. J Biol Chem 266:20369–20374
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng J, Behringer R, de Crombrugghe B (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108:17–29
Nakashima K, de Crombrugghe B (2003) Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet 19:458–466
Neumann E, Schett G (2007) Bone metabolism: molecular mechanisms. Z Rheumatol 66:286–289
O'Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T (2008) Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 3:e2942
Odgren PR, Kim N, MacKay CA, Mason-Savas A, Choi Y, Marks SC Jr (2003) The role of RANKL (TRANCE/TNFSF11), a tumor necrosis factor family member, in skeletal development: effects of gene knockout and transgenic rescue. Connect Tissue Res 44:264–271
Phimphilai M, Zhao Z, Boules H, Roca H, Franceschi RT (2006) BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J Bone Miner Res 21:637–646
Piters E, Boudin E, Van Hul W (2008) Wnt signaling: a win for bone. Arch Biochem Biophys 473:112–116
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Löwik CW, Reeve J (2005) Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J 19:1842–1844
Ramasamy I (2006) Recent advances in physiological calcium homeostasis. Clin Chem Lab Med 44:237–273
Roodman GD (2006) Regulation of osteoclast differentiation. Ann N Y Acad Sci 1068:100–109
Saftig P, Hunziker E, Everts V, Jones S, Boyde A, Wehmeyer O, Suter A, von Figura K (2000) Functions of cathepsin K in bone resorption. Lessons from cathepsin K deficient mice. Adv Exp Med Biol 477:293–303
Silver IA, Murrills RJ, Etherington DJ (1988) Microelectrode studies on the acid microenvironment beneath adherent macrophages and osteoclasts. Exp Cell Res 175:266–276
Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, Shimamoto G, DeRose M, Elliott R, Colombero A, Tan HL, Trail G, Sullivan J, Davy E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 20:345–357
Takahashi N, Udagawa N, Suda T (1999) A new member of tumor necrosis factor ligand family, ODF/OPGL/TRANCE/RANKL, regulates osteoclast differentiation and function. Biochem Biophys Res Commun 256:449–455
Takahashi N, Akatsu T, Udagawa N, Sasaki T, Yamaguchi A, Moseley JM, Martin TJ, Suda T (1988) Osteoblastic cells are involved in osteoclast formation. Endocrinology 123:2600–2602
Teitelbaum SL (2000) Bone resorption by osteoclasts. Science 289:1504–1508
Theill LE, Boyle WJ, Penninger JM (2002) RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu Rev Immunol 20:795–823
Tolar J, Teitelbaum SL, Orchard PJ (2004) Osteopetrosis. N Engl J Med 351:2839–2849
Urist MR (1965) Bone: formation by autoinduction. Science 150:893–899
van Bezooijen R, Dijke P, Papapoulos S, Löwik CGM (2005) Sclerostin, an osteocyte-derived negative regulator of bone formation. Cytokine Growth Factor Rev 16:319–327
Väänänen HK, Laitala-Leinonen T (2008) Osteoclast lineage and function. Arch Biochem Biophys 473:132–138
Wada T, Nakashima T, Hiroshi N, Penninger JM (2006) RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med 12:17–25
Wennberg C, Hessle L, Lundberg P, Mauro S, Narisawa S, Lerner UH, Millán JL (2000) Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J Bone Miner Res 15:1879–1888
Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, Tsuda E, Morinaga T, Higashio K, Udagawa N, Takahashi N, Suda T (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 95:3597–3602
Yavropoulou MP, Yovos JG (2007) The role of the Wnt signaling pathway in osteoblast commitment and differentiation. Hormones (Athens) 6:279–294
Yoshida H, Hayashi S, Kunisada T, Ogawa M, Nishikawa S, Okamura H, Sudo T, Shultz LD, Nishikawa S (1990) The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 345:442–444
Acknowledgement
The authors acknowledge the expert technical assistance of Simone Thiemer (Department of Orthodontics, Regensburg University) for designing the figures.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Proff, P., Römer, P. The molecular mechanism behind bone remodelling: a review. Clin Oral Invest 13, 355–362 (2009). https://doi.org/10.1007/s00784-009-0268-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00784-009-0268-2