
Abstract
The skeleton is a dynamic tissue system comprised of cancellous and cortical bone that is constantly being remodeled throughout postnatal life. During normal physiological conditions, bone mass and integrity are maintained through bone resorbing osteoclasts, and bone forming osteoblasts. Bone remodeling takes place in the Haversian system through the coordinated effort of basic multicellular units, such as osteoclasts, osteoblasts, and mechanosensitive osteocytes. Osteocytes, the most abundant bone cell type, can also directly control bone mass via peri-lacunar remodeling. Local and systemic factors play an important role in regulating basic multicellular unit function either by promoting, or inhibiting, osteoclast or osteoblast maturation and activity. These local factors and mechanisms controlling bone formation, particularly via osteocytes, include gap junctional intercellular communication, RANKL-OPG secretion and WNT/β-catenin signaling. Systemic factors that have actions on multiple bone cell types include parathyroid hormone and estrogen. Understanding how these biomolecules influence bone cells and control bone remodeling is critical to developing treatments to prevent skeletal diseases, such as osteoporosis, and resulting fracture.
The present invited review was completed and submitted to the publisher on 21-Feb-20.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Osteoblast
- Osteoclast
- Osteocytes
- Cancellous
- Cortical
- Haversian
- Basic multicellular unit
- Perilacunar
- Bone remodeling
1 Clinical Rationale
1.1 Introduction
Bone is a dynamic mixture of cells, collagen and hydroxyapatite (Ca10(PO4)6(OH)2). In order to maintain its structural integrity and provide mineral homeostasis bone is constantly remodeled throughout postnatal life [1]. Normal bone remodeling occurs in either bone type (cortical or cancellous) by the coordinated action of bone formation by osteoblasts and bone resorption by osteoclasts. In normal physiological conditions, bone formation and resorption are nearly equal and there is no net change in bone mass. However, under different levels of exercise or pathological conditions bone mass can change due to an imbalance in overall remodeling. When bone mass decreases, skeletal fragility can result, which subsequently increases a patient’s risk of fracture. The increased risk of fracture is only partially explained by a reduction in bone mass and is dictated by other parameters such as bone structure and material quality [2]. Nonetheless, increased skeletal fragility is found in patients with bone remodeling disorders such as osteoporosis and other pathologies [3, 4].
1.2 Bone Remodeling Disorders
Osteoporosis is a disease characterized by low bone mass, compromised bone strength, deterioration of bone tissue, and disrupted bone architecture. It has a widespread incidence affecting 1 in 2 women and 1 in 5 men over the age of 50 [5]. The clinical definition of osteoporosis, defined by the World Health Organization (WHO), is bone mineral density (BMD) at the hip or lumbar spine of a patient being less than or equal to 2.5 standard deviations below the young healthy reference population (30 years old) mean BMD (called a T-score) [6]. Osteopenia is a disease in which bone density is below average (T-score between −1 and −2.5), though not as severe as osteoporosis [7]. Some causes of low bone mass are estrogen deficiency, as seen in postmenopausal women, glucocorticoids, disuse, and diabetes/obesity [6]. Treatments to alleviate symptoms of osteoporosis include lifestyle changes, such as cessation of smoking, reduction of alcohol use, and increased physical activity. Vitamin D and calcium supplements may also improve bone health but can have unintended drawbacks seen in other organ systems, such as calcium buildup in the cardiovascular system [8]. The most effective pharmaceutical therapies for osteoporosis treatment are anti-resorption drugs, such as bisphosphonates (Chapter “Effects of Osteoporosis Drugs–Morphological Assessment and Adverse Events”) and Denosumab (Chapter “Denosumab in the Treatment of Postmenopausal Women with Osteoporosis: Fracture Outcomes, BMD, and Morphological Assessment”), that minimize bone resorption [8,9,10].
2 Bone Cells and Associated Functions (Fig. 1)
Osteoclasts are multinucleated cells, derived from monocytes of the hematopoietic lineage that function to resorb bone. The differentiation of monocyte lineage cells into mature osteoclasts is dependent on receptor activator of NF-kB ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) [11]. M-CSF binds to its receptor (c-Fms) on osteoclast precursors and mature cells to stimulate their proliferation and survival [11]. However, RANKL, which is expressed by both osteoblasts and osteocytes, is the primary osteoclast differentiation factor [11]. RANKL binds to its receptor, RANK, present on osteoclasts, to induce transcription factors and enzymes that promote bone resorption. Osteoprotegerin (OPG), an endogenous antagonist to RANKL, inhibits RANKL binding to RANK by acting as a decoy receptor [8, 12]. OPG is secreted by osteoblast lineage cells to aid in the tight regulation of osteoclastogenesis [12, 13]. Osteoclasts are able to resorb bone due to their motile cytoskeleton, adhesion molecules, and a ruffled border at the bone surface [8, 14]. These qualities of osteoclasts allow for their attachment to bone, largely via αVβ3 integrins, and the creation of a sealed-off microenvironment between the osteoclast ruffled border and the bone surface. At the ruffled border active carbonic anhydrase converts CO2 and H2O to bicarbonate (HCO3-) and protons (H+) that are secreted into the resorption compartment creating an acidic environment. This acidic environment allows for activation of osteoclast specific matrix metalloproteinases, including tartrate resistant acid phosphatase (TRAP), and the collagenase cathepsin K, which facilitates breakdown of the extracellular matrix and resorption of bone matrix [8, 15, 16]. Cathepsin K aids in removing poor quality bone where microcracks have accumulated and hole-like lacunae have formed [8]. TRAP is highly expressed by mature osteoclasts during bone resorption and is critical for skeletal development [14, 17]. Another highly expressed enzyme produced by mature osteoclasts is Src kinase [8]. Src kinase’s main role is to mediate multiple pathways that regulate osteoclast activity, but not number, such as formation of a ruffled border critical for bone resorption. Without the presence of Src kinase there will be an increase in osteoblastic bone formation, but no effect on overall osteoclast number.

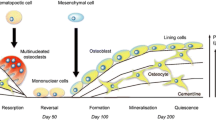
Overview of cellular and molecular mediators of bone remodeling. The Bone Remodeling Compartment (BRC) is a specialized environment where bone remodeling occurs through osteoclasts resorbing old or damaged bone tissue, and osteoblasts forming new bone matrix. Bone remodeling is tightly regulated and is driven by local osteocytic factors such as RANKL, OPG, and WNT ligands or via endocrine signaling by estrogen and PTH. Specifically, estrogen acts on osteoblasts and osteocytes to promote bone formation, while inhibiting osteoclastic bone resorption. PTH has pleiotropic effects on bone. It acts on the osteoblasts, osteocytes, and bone lining cells to directly promote bone formation or to cause osteoblasts and osteocytes to release RANKL. RANKL then binds to RANK receptors on osteoclasts, to promote osteoclastogenesis. However, osteoblasts and osteocytes release a decoy receptor, OPG, that tightly regulates osteoclastogenesis by inhibiting RANKL-RANK binding. WNT ligands, part of the β-catenin canonical signaling pathway, play a major role in bone remodeling by acting on osteoblasts and osteocytes to promote osteoblastogenesis and indirectly control osteoclastogenesis. Abbreviations: receptor activator of NF-κB ligand, OPG, osteoprotegerin, PTH, parathyroid hormone
Osteoblasts, derived from mesenchymal stem cells, are primarily responsible for the synthesis of bone extracellular matrix. This process, often referred to as osteogenesis, is crucial for the proper development and maintenace of bone. In adult skeletons, osteoblasts are recruited to regenerate, or remodel, areas of bone with depleted matrix or defects [8, 18]. To synthesize bone at sites of resorption, osteoblasts secrete bone matrix proteins, such as collagen type 1 (Col1) and noncollagenous proteins such as osteocalcin (OCN) and osteopontin (OPN). Through osteoblast derived alkaline phosphatase (ALP) [8, 19] the synthesized bone extracellular matrix (called osteoid) undergoes mineralization to form hydroxyapatite [18,19,20]. Osteoblast differentiation is controlled by two well-studied transcription factors: Runt-related transcription factor 2 (RUNX2) and Osterix (Osx). RUNX2 is an essential bone-specific transcription factor that causes the upregulation of osteoblast differentiation marker genes and induces osteogenesis [8, 21]. Osx is located downstream of RUNX2 and is required for osteoblast maturation and bone mineralization [18, 21, 22]. Osteoblast growth and differentiation are also mediated by different classical signaling pathways such as transforming growth factor-beta (TGF-β), bone morphogenetic proteins (BMPs), fibroblast growth factors (FGFs), and the WNT/ β -catenin pathway [23]. Furthermore, osteoblasts are sensitive to mechanical stimuli resulting from mechanical loading [8, 18, 20].
Osteocytes are the most abundant cell type in bone and comprise about 90–95% of all the cells in bone [24]. They are terminally differentiated, post-mitotic osteoblasts that have become embedded in mineralized bone matrix. The osteocyte has two major components: (1) a central cell body in a bone cavity called a lacuna (15–20 microns in humans) and (2) small dendritic projections (50–100 per cell) [25] coming off the cell body that protrude through microscopic (50–300 nm) [26] channels called canaliculi. This vast network of lacunae and canaliculi make up the lacunar-canalicular system of bone, which is thought to serve as a biochemical transport system between osteocytes, vascular channels, and bone surfaces as well as a mechanical amplification system. Through these expansive networks, osteocytes can connect and transport small molecules to neighboring bone cells via gap junctions (see section “Important Molecules in Bone Remodeling”). Due to these features, osteocytes have been implicated as the key mechanosensitive and endocrine regulated cell in bone that regulates bone mass. Osteocytes express higher levels of key bone matrix and phosphate regulatory molecules, including osteocalcin (OCN, dentin-matrix protein 1 (DMP1), phosphate-regulating neutral endopeptidase (PHEX), matrix extracellular, phosphoglycoproteine (MEPE) and fibroblast growth factor 23 (FGF23), than osteoblasts [27]. They also express molecules that directly inhibit osteoblast formation and activity, such as dickkopf-related protein 1 (Dkk1) and sclerostin, and regulate osteoclast formation via RANKL and its decoy receptor OPG. Osteocytes can live the entire lifetime of the host (mean half-life 25 years) [28] or undergo apoptosis (i.e., programmed cell death) due to various environmental stimuli such as bone microdamage (linear microcracks), estrogen deficiency, unloading, or glucocorticoid use [29]. It was first shown by Cardoso et al. that osteocyte apoptosis, which occurs in microdamaged regions of the bone following fatigue loading (which results in microcracks), is a key event necessary for targeted intracortical remodeling [30]. Subsequent in vitro and in vivo work has suggested that apoptotic osteocytes near fatigue damage microcracks in bone (≤300 microns) do not secrete any signals and therefore this response is fairly targeted to the damaged bone region. This same effect is apparent in estrogen withdrawal after ovariectomy, where osteocyte apoptosis is upregulated 4–7 times in the posterior diaphyseal cortex, resulting in activation of endosteal resorption [33]. Estrogen signaling is necessary for continued mitigation of mitogen-activated protein kinases (MAPKs) activation, which neutralizes reactive oxygen species (ROS) in osteocytes thereby preventing apoptosis [29].
2.1 Bone Types and Haversian System
Structurally, bone is composed of two distinct types, cancellous and cortical. Cancellous bone is found at the end of long bones (epiphysis/metaphysis) or in flat bones, such as the pelvis, clavicle and cranium, and is made up of highly arranged trabecular struts. These trabecular struts, in humans, can be dominated by a rod- or plate-like architecture, are 150–300 microns thick and interspaced every 0.5 to 1.5 mm [34, 35]. Therefore, cancellous bone has a large surface area to volume ratio (porosity of 0.5–0.95) and its remodeling serves a primary metabolic function in the body. Cancellous bone remodeling starts at the surface of the trabeculae and usually takes about 200 days to complete [36]. In contrast, Cortical bone, which is denser and more compact, makes up the outer covering of all bone and is sometimes referred to as compact bone. Cortical bone’s structure and a usually lower rate of turnover enable it to serve a primarily load-bearing function. Human cortical bone is initially formed and made up of discrete units called osteons that make up the Haversian system of bone. These osteons are cylindrical in nature and consist of blood vessels and nerves at their center (Haversian canal) surrounded by rings of concentric layers of compact bone called lamellae. Interspersed between these layers of lamellar bone are osteocytes and their lacunar-canalicular system. Osteons are typically 200 μm in diameter and 3 mm in length [37]. Both cancellous and osteonal remodeling occurs by the stochastic and coordinated action of multiple cell types, termed basic multicellular units (BMU’s), that remodels bone [38, 39].
2.2 “Basic Multicellular Unit” BMU-Based Remodeling
The replacement of old osteons in cortical bone or trabecular struts to form secondary (i.e. new) osteons or new trabeculae, respectively, occurs by the coordinated action of a group of bone cells termed the “Basic Multicellular Unit” (BMU), which creates a structural feature of bone tissue called the “Bone Remodeling Unit” (BRU) [40]. These processes occur in a specialized environment, that is enclosed by canopy cells and is vascularized and innervated, called the “Bone Remodeling Compartment” (BRC) [41] (see Chapter “Cellular and Molecular Biology in Bone Remodeling”; Fig. 1 and Chapter “Mechanism Reversing Bone Resorption to Formation During Bone Remodeling”). Bone remodeling in total occurs across several discrete phases named in order of their occurrence; Activation-Resorption/Reversal-Formation (A-R-F) (see Chapter “Bone Remodeling and Modeling: Therapeutic Targets for the Treatment of Osteoporosis”). In the activation phase, hormonal (PTH) or mechanical remodeling signals (RANKL, microdamage) sensed by the osteocyte and osteoblasts, signal to pre-osteoclasts to undergo osteoclast formation and resorptive activity [42,43,44]. This activation phase is quick and lasts 1–3 days. Once osteoclasts have formed, the resorption phase begins with the osteoclasts forming an ellipsoidal “cutting cone” in cortical bone or Howship’s lacunae in cancellous bone [45]. The osteoclasts begin to resorb bone along the anatomical axis of the osteon or trabeculae at a rate of 20–40 μm per day for about 3 weeks [46], leaving behind demineralized and partially digested bone matrix. Following osteoclast-mediated resorption, a reversal stage begins that initiates bone formation (see Chapter “Significance of Reversal Resorption Phase in Bone Loss”). Although the cell type responsible for initiating bone formation is contested [1, 47,48,49], it is generally accepted that this specialized cell prepares the bone surface and potentially secretes coupling factors to promote osteoblast formation and activation. In the last phase of BMU based bone remodeling, the formation phase, osteoblasts deposit a new unmineralized bone matrix, called osteoid, into the prepared resorption spaces. This osteoid is later mineralized with the help of specialized metalloenzymes, such as alkaline phosphatase, from the osteoblast [50, 51]. These osteoblasts can then either become entrapped in minerals to form osteocytes, undergo apoptosis, or become quiescent to form lining cells that cover the new bone surface to aid in the next remodeling cascade [52, 53].
2.3 Perilacunar Remodeling
The lacunar-canalicular system is vast. In humans, it makes up an area of bone nearly 215 m2 [2] and therefore represents a large reservoir of available minerals and bioactive molecules. Osteocytic osteolysis, or the newer term, perilacunar remodeling (also called perilacunar turnover to distinguish it from bone remodeling involving the BMU which requires both osteoclasts and osteoblasts), is now recognized as a distinct form of bone turnover that is carried out directly by the osteocyte. Typically, perilacunar remodeling is associated with conditions that place rapid demands on the skeleton, such as lactation, calcium restriction, and space flight. In fact, experiments have shown that at any one time 15–20% of osteocytes are associated with new bone-forming surfaces and can produce the same enzymes used by osteoclasts to resorb bone including matrix metalloproteases (MMPs), proton pumps, carbonic anhydrase, and cathepsin K [24, 54, 55]. More recent evidence has shown that perilacunar remodeling is coordinated via TGFΒ-β and PTH receptor signaling in osteocytes as well as the downstream transcriptional regulators Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) [54, 56, 57]. More importantly, these same studies show that perilacunar remodeling has a direct effect on the local bone matrix quality and therefore can directly influence fracture resistance.
3 Important Molecules in Bone Remodeling
3.1 Local (Paracrine and Autocrine)
Receptor activator of nuclear factor-kappa-β ligand (RANKL), its receptor RANK and Osteoprotegerin (OPG)—RANKL is expressed in the local bone microenvironment by osteoblast lineage cells (osteoblasts and osteocytes) and signals to its receptor RANK on osteoclastic precursor cells to control their fusion, survival, and differentiation into mature osteoclasts [58]. RANKL is highly upregulated in bone cells, especially osteocytes exposed to PTH, after bone damage, and disuse [59]. People with mutations in the RANKL gene (TNFSF11) demonstrate osteopetrosis or excessively brittle bone due to hypermineralization [60]. On the other hand, OPG is a decoy receptor for RANKL that is expressed by osteoblast lineage cells and blocks osteoclastogenesis through competitive inhibition of RANKL-RANK binding [61]. OPG is upregulated due to bone loading, estrogen, and transforming growth factor β (TGF-β) [62]. Therefore, RANKL/OPG pathways play a key role in coordinating bone resorption by various local and systemic regulators.
WNT/β-Catenin Signaling and Sclerostin—The WNT/β-catenin signaling pathway has emerged as a major pathway controlling bone remodeling. In canonical WNT signaling, WNT ligands (19 distinct proteins in vertebrates) bind to the frizzled receptor with co-activators LRP4/5/6. This results in β-catenin stabilization, nuclear translocation and binding to TCF/LEF in the cell nucleus. This stabilization increases transcriptional activation of genes that increase osteocyte/osteoblast proliferation, viability, and bone formation while simultaneously decreasing bone resorption via decreased RANKL/OPG signaling [63, 64]. Therefore, modulating WNT/β-catenin has become a key target to improve bone mass and quality by increasing bone formation while downregulating resorption. One key approach used to modulate WNT/β-Catenin signaling is by inhibiting sclerostin, the product of the SOST gene and a WNT/β-Catenin antagonist that binds LRP5/6. Sclerostin is primarily produced by osteocytes, is regulated by loading, and mutations in the gene lead to high bone mass disorders such as Sclerosteosis or Van Buchem’s disease [65,66,67,68].
Gap Junctional Intercellular Communication (GJIC)—Gap junctions are membrane-spanning protein channels that allow for the passage of small molecules such as ATP, calcium, prostaglandin, and microRNAs between bone cells [69]. The predominate gap junction protein in bone is connexin 43 (Cx43), which is encoded by the GJA1 gene and highly expressed by osteoblast lineage cells, especially osteocytes. It is found at the connections between osteocytes and other bone cells and is necessary for osteocytic regulation of osteoblasts via mechanical stimulation [70]. Mutations in the GJA1 gene in humans cause oculodentodigital dysplasia and are associated with high turnover osteopenia and increased intracortical porosity [71,72,73]. This is likely because Cx43 is necessary for normal osteoblast lineage cell differentiation, development, and function. Cx43 has also been shown to play a large role in skeletal homeostasis and skeletal adaptation to mechanical loading. For example, loss of Cx43 in osteoblast lineage cells (osteoblasts and osteocytes) attenuates bone loss seen with unloading [74, 75]. Interestingly, the same conditional deletion of Cx43 leads to greater bone formation with skeletal loading [76, 77]. These studies and recent reviews suggest that these beneficial skeletal results are due to Cx43’s regulation of osteocyte apoptosis and specific factors such as PGE2, sclerostin, and RANKL/OPG, respectively, that are critical for normal bone remodeling [74, 78,79,80,81].
3.2 Systemic (Endocrine)
Parathyroid Hormone (PTH) is an important hormone for maintaining bone homeostasis. It is secreted by the parathyroid glands and plays a key role in coordinating serum calcium and phosphate homeostasis [82]. Specifically, PTH works by binding to its receptor, which is found on all cells of the osteoblast lineage (i.e., bone lining cells, osteoblasts, osteocytes) [83]. PTH receptor activation increases bone formation by converting bone lining cells to active osteoblasts, decreasing osteoblast apoptosis and promoting osteoblastic differentiation by acting as an upstream regulator of RUNX2 [43, 84]. Furthermore, PTH receptor activation of osteocytes increases RANKL mediated osteoclastogenesis and decreases sclerostin expression [85, 86]. However, the effect of PTH on bone is time-dependent. While a constant, elevated level of PTH stimulates bone resorption, an intermittent dose is anabolic and promotes bone formation [18]. Teriparatide, a recombinant form of active PTH, is the oldest and most commonly used bone anabolic drug to treat osteoporosis [87] (see Chapter “Teriparatide”).
Estrogen (17β-estradiol) is another important hormone for bone, particularly for women. 17β-estradiol, the most common form of estrogen, has direct effects on bone cells by binding to the estrogen receptors alpha and or beta (ERα and ERβ), which are found on osteoblasts, osteoclasts, and osteocytes [88]. ERα and ERβ activation directly promotes osteocyte and osteoblast function, inhibits their apoptosis, and inhibits osteoclast activation and function [88,89,90]. All of these estrogen functions work to prevent bone loss and maintain skeletal mineralization. However, postmenopausal women experience a decline in ovarian estradiol levels, which leads to decreased BMD levels [89, 91]. Men also experience a decline of estrogen levels with age, but not to the same extent as women [89]. These decreased serum estradiol levels can be predictive of bone fracture and development of osteoporosis in both men and women [89, 91].
Other systemic factors, including but not limited to Calcitonin, FGF23, and Insulin are also important for bone remodeling. Calcitonin is released by thyroid cells and binds to osteoclasts to inhibit bone resorption [92]. FGF23 is produced by mature osteocytes to help the kidneys regulate serum phosphorous levels and maintain mineral homeostasis [93,94,95]. Insulin is an important hormone in both energy and bone metabolism [96]. Insulin is recruited to bone through the osteoblastic release of osteocalcin, which signals to pancreatic β-cells to release insulin [97]. Insulin acts directly on bone cells by decreasing OPG production from osteoblasts, thereby increasing RANKL concentrations that can activate osteoclasts [97].
Biomarkers and Assays of Bone Remodeling—To determine the status of bone turnover, serum biochemical markers, and bone biopsies are used clinically (see Chapter “Metabolic Boneturnover Markers”). The iliac crest is a common biopsy site used to simultaneously observe cortical and trabecular bone mineralization and cellular features histologically [98, 99]. Usually, iliac crest biopsies are taken after systemic administration of a bone chelating agent, such as tetracycline, which can be analyzed histologically to assess rates of bone turnover [98,99,100] (see Chapter “Histomorphometric Assessment of Remodeling and Modeling-Based Bone Formation ”). A noninvasive assay to evaluate bone turnover involves quantifying biochemical markers in blood serum following fasting. Common serum bone formation markers are osteocalcin (OCN) and procollagen type I N-terminal propeptide (P1NP). Common serum bone resorption markers are C-terminal telopeptide (CTX), N-terminal telopeptide (NTX), and TRAP5b [98, 101]. Other serum biochemical markers assessed include PTH, insulin-like growth factor I (IGF-I) (an anabolic hormone for bone), and vitamin D [96, 101].
References
Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285(33):25103–8.
Fonseca H, et al. Bone quality: the determinants of bone strength and fragility. Sports Med. 2014;44(1):37–53.
Unnanuntana A, et al. The assessment of fracture risk. J Bone Joint Surg Am. 2010;92(3):743–53.
Cauley JA. Public health impact of osteoporosis. J Gerontol A Biol Sci Med Sci. 2013;68(10):1243–51.
Russell RG, Espina B, Hulley P. Bone biology and the pathogenesis of osteoporosis. Curr Opin Rheumatol. 2006;18(Suppl 1):S3–10.
Cosman F, et al. Clinician’s guide to prevention and treatment of osteoporosis. Osteoporos Int. 2014;25(10):2359–81.
Karaguzel G, Holick MF. Diagnosis and treatment of osteopenia. Rev Endocr Metab Disord. 2010;11(4):237–51.
Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: now and the future. Lancet (London, England). 2011;377(9773):1276–87.
Baron R, Ferrari S, Russell RG. Denosumab and bisphosphonates: different mechanisms of action and effects. Bone. 2011;48(4):677–92.
Ominsky MS, et al. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011;26(5):1012–21.
Feng X, Teitelbaum SL. Osteoclasts: new insights. Bone Res. 2013;1(1):11–26.
Baud'huin M, et al. Osteoprotegerin: multiple partners for multiple functions. Cytokine Growth Factor Rev. 2013;24(5):401–9.
Fu YX, et al. Osteoprotegerin influences the bone resorption activity of osteoclasts. Int J Mol Med. 2013;31(6):1411–7.
Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–42.
Henriksen K, et al. Osteoclast activity and subtypes as a function of physiology and pathology--implications for future treatments of osteoporosis. Endocr Rev. 2011;32(1):31–63.
Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289(5484):1504–8.
Blumer MJF, et al. Role of tartrate-resistant acid phosphatase (TRAP) in long bone development. Mech Dev. 2012;129(5):162–76.
Rutkovskiy A, Stensløkken K-O, Vaage IJ. Osteoblast differentiation at a glance. Med Sci Monit Basic Res. 2016;22:95–106.
Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2011;13(1):27–38.
Neve A, Corrado A, Cantatore FP. Osteoblast physiology in normal and pathological conditions. Cell Tissue Res. 2011;343(2):289–302.
Vimalraj S, et al. Runx2: structure, function, and phosphorylation in osteoblast differentiation. Int J Biol Macromol. 2015;78:202–8.
Zhang C. Transcriptional regulation of bone formation by the osteoblast-specific transcription factor Osx. J Orthop Surg Res. 2010;5:37.
Huang W, et al. Signaling and transcriptional regulation in osteoblast commitment and differentiation. Front Biosci. 2007;12:3068–92.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–38.
Beno T, et al. Estimation of bone permeability using accurate microstructural measurements. J Biomech. 2006;39(13):2378–87.
Han Y, et al. Mechanotransduction and strain amplification in osteocyte cell processes. Proc Natl Acad Sci U S A. 2004;101(47):16689–94.
Bellido T. Osteocyte-driven bone remodeling. Calcif Tissue Int. 2014;94(1):25–34.
Dallas SL, Prideaux M, Bonewald LF. The osteocyte: an endocrine cell … and more. Endocr Rev. 2013;34(5):658–90.
Jilka RL, Noble B, Weinstein RS. Osteocyte apoptosis. Bone. 2013;54(2):264–71.
Cardoso L, et al. Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue. J Bone Miner Res. 2009;24(4):597–605.
McCutcheon S, et al. Apoptotic osteocytes induce RANKL production in bystanders via purinergic signaling and activation of pannexin channels. J Bone Miner Res. 2020;35(5):966–77.
Kennedy OD, et al. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone. 2014;64:132–7.
Emerton KB, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46(3):577–83.
Croucher PI, Garrahan NJ, Compston JE. Assessment of cancellous bone structure: comparison of strut analysis, trabecular bone pattern factor, and marrow space star volume. J Bone Miner Res. 1996;11(7):955–61.
Hildebrand T, et al. Direct three-dimensional morphometric analysis of human cancellous bone: microstructural data from spine, femur, iliac crest, and calcaneus. J Bone Miner Res. 1999;14(7):1167–74.
Eriksen EF. Cellular mechanisms of bone remodeling. Rev Endocr Metab Disord. 2010;11(4):219–27.
Britz HM, et al. The relation of femoral osteon geometry to age, sex, height and weight. Bone. 2009;45(1):77–83.
Frost HM. Treatment of osteoporoses by manipulation of coherent bone cell populations. Clin Orthop Relat Res. 1979;143:227–44.
Frost HM. Bone “mass” and the “mechanostat”: a proposal. Anat Rec. 1987;219(1):1–9.
Frost HM. Bone remodelling dynamics. Thomas; 1963.
Hauge EM, et al. Cancellous bone remodeling occurs in specialized compartments lined by cells expressing osteoblastic markers. J Bone Miner Res. 2001;16(9):1575–82.
Powell WF, et al. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol. 2011;209(1):21–32.
Kim SW, et al. Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res. 2012;27(10):2075–84.
Kennedy OD, et al. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50(5):1115–22.
Patterson-Kane JC, Firth EC. Chapter 13 - Tendon, ligament, bone, and cartilage: anatomy, physiology, and adaptations to exercise and training. In: Hodgson DR, McKeever KH, McGowan CM, editors. The athletic horse. 2nd ed. W.B. Saunders; 2014. p. 202–42.
Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci. 2006;1092:385–96.
Pettit AR, et al. Osteal macrophages: a new twist on coupling during bone dynamics. Bone. 2008;43(6):976–82.
Raisz LG. Physiology and pathophysiology of bone remodeling. Clin Chem. 1999;45(8 Pt 2):1353–8.
Delaisse JM. The reversal phase of the bone-remodeling cycle: cellular prerequisites for coupling resorption and formation. Bonekey Rep. 2014;3:561.
Blair HC, et al. Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Eng Part B Rev. 2017;23(3):268–80.
Golub EE, Boesze-Battaglia K. The role of alkaline phosphatase in mineralization. Curr Opin Orthop. 2007;18(5):444–8.
Maes C, Kobayashi T, Kronenberg HM. A novel transgenic mouse model to study the osteoblast lineage in vivo. Ann N Y Acad Sci. 2007;1116:149–64.
Clarke B. Normal bone anatomy and physiology. Clin J Am Soc Nephrol. 2008;3(Suppl 3):S131–9.
Qing H, et al. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27(5):1018–29.
Yee CS, et al. Investigating osteocytic perilacunar/canalicular remodeling. Curr Osteoporos Rep. 2019;17(4):157–68.
Dole NS, et al. Osteocyte-intrinsic TGF-β signaling regulates bone quality through perilacunar/canalicular remodeling. Cell Rep. 2017;21(9):2585–96.
Kegelman CD, et al. YAP and TAZ mediate osteocyte perilacunar/canalicular remodeling. J Bone Miner Res. 2019.
Kong YY, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–23.
Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473(2):139–46.
Sobacchi C, et al. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9(9):522–36.
Lacey DL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–76.
Theoleyre S, et al. The molecular triad OPG/RANK/RANKL: involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004;15(6):457–75.
Duan P, Bonewald LF. The role of the wnt/β-catenin signaling pathway in formation and maintenance of bone and teeth. Int J Biochem Cell Biol. 2016;77(Pt A):23–9.
Kobayashi Y, Maeda K, Takahashi N. Roles of Wnt signaling in bone formation and resorption. Jpn Dental Sci Rev. 2008;44(1):76–82.
Robling AG, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–75.
Balemans W, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet. 2001;10(5):537–43.
Brunkow ME, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–89.
Lin C, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J Bone Miner Res. 2009;24(10):1651–61.
Donahue HJ, Qu RW, Genetos DC. Joint diseases: from connexins to gap junctions. Nat Rev Rheumatol. 2017;14:42.
Taylor AF, et al. Mechanically stimulated osteocytes regulate osteoblastic activity via gap junctions. Am J Physiol Cell Physiol. 2007;292(1):C545–52.
Flenniken AM, et al. A Gja1 missense mutation in a mouse model of oculodentodigital dysplasia. Development. 2005;132(19):4375–86.
Lloyd SA, et al. Shifting paradigms on the role of connexin43 in the skeletal response to mechanical load. J Bone Miner Res. 2014;29(2):275–86.
Lloyd SA, et al. Evidence for the role of connexin 43-mediated intercellular communication in the process of intracortical bone resorption via osteocytic osteolysis. BMC Musculoskelet Disord. 2014;15(1):122.
Lloyd SA, et al. Connexin 43 deficiency attenuates loss of trabecular bone and prevents suppression of cortical bone formation during unloading. J Bone Miner Res. 2012;27(11):2359–72.
Lloyd SA, et al. Connexin 43 deficiency desensitizes bone to the effects of mechanical unloading through modulation of both arms of bone remodeling. Bone. 2013;57(1):76–83.
Bivi N, et al. Absence of Cx43 selectively from osteocytes enhances responsiveness to mechanical force in mice. J Orthop Res. 2013;31(7):1075–81.
Zhang Y, et al. Enhanced osteoclastic resorption and responsiveness to mechanical load in gap junction deficient bone. PLoS One. 2011;6(8):e23516.
Xu H, et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J Bone Miner Res. 2015;30(3):436–48.
Ma L, et al. Connexin 43 hemichannels protect bone loss during estrogen deficiency. Bone Res. 2019;7:11.
Klein-Nulend J, et al. Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts--correlation with prostaglandin upregulation. Biochem Biophys Res Commun. 1995;217(2):640–8.
Genetos DC, et al. Oscillating fluid flow activation of gap junction hemichannels induces atp release from MLO-Y4 osteocytes. J Cell Physiol. 2007;212(1):207–14.
Gensure RC, Gardella TJ, Jüppner H. Parathyroid hormone and parathyroid hormone-related peptide, and their receptors. Biochem Biophys Res Commun. 2005;328(3):666–78.
Wein MN, Kronenberg HM. Regulation of bone remodeling by parathyroid hormone. Cold Spring Harb Perspect Med. 2018;8(8):a031237.
Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40(6):1434–46.
Xiong J, et al. Osteocyte-derived RANKL is a critical mediator of the increased bone resorption caused by dietary calcium deficiency. Bone. 2014;66:146–54.
Spatz JM, et al. The Wnt inhibitor sclerostin is up-regulated by mechanical unloading in osteocytes in vitro. J Biol Chem. 2015;290(27):16744–58.
Eastell R, Walsh JS. Anabolic treatment for osteoporosis: teriparatide. Clin Cases Miner Bone Metab. 2017;14(2):173–8.
Khalid AB, Krum SA. Estrogen receptors alpha and beta in bone. Bone. 2016;87:130–5.
Cauley JA. Estrogen and bone health in men and women. Steroids. 2015;99(Pt A):11–5.
Khosla S, Oursler MJ, Monroe DG. Estrogen and the skeleton. Trends Endocrinol Metab. 2012;23(11):576–81.
Carson JA, Manolagas SC. Effects of sex steroids on bones and muscles: similarities, parallels, and putative interactions in health and disease. Bone. 2015;80:67–78.
Keller J, et al. Calcitonin controls bone formation by inhibiting the release of sphingosine 1-phosphate from osteoclasts. Nat Commun. 2014;5:5215.
Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol. 2007;18(6):1637–47.
Sapir-Koren R, Livshits G. Osteocyte control of bone remodeling: is sclerostin a key molecular coordinator of the balanced bone resorption-formation cycles? Osteoporos Int. 2014;25(12):2685–700.
Clinkenbeard EL, et al. Increased FGF23 protects against detrimental cardio-renal consequences during elevated blood phosphate in CKD. JCI Insight. 2019;4(4).
Curtis E, et al. Determinants of muscle and bone aging. J Cell Physiol. 2015;230(11):2618–25.
Clemens TL, Karsenty G. The osteoblast: an insulin target cell controlling glucose homeostasis. J Bone Miner Res. 2011;26(4):677–80.
Cohen A, et al. Abdominal fat is associated with lower bone formation and inferior bone quality in healthy premenopausal women: a transiliac bone biopsy study. J Clin Endocrinol Metab. 2013;98(6):2562–72.
Priemel M, et al. Bone mineralization defects and vitamin D deficiency: histomorphometric analysis of iliac crest bone biopsies and circulating 25-hydroxyvitamin D in 675 patients. J Bone Miner Res. 2010;25(2):305–12.
Hernandez JD, et al. Technical approach to iliac crest biopsy. Clin J Am Soc Nephrol. 2008;3(Suppl 3):S164–9.
Chavassieux P, et al. Are biochemical markers of bone turnover representative of bone histomorphometry in 370 postmenopausal women? J Clin Endocrinol Metab. 2015;100(12):4662–8.
Acknowledgments
This work is supported by the Translational Research Institute for Space Health through NASA cooperative agreement NNX16A069A, National Institutes of Health (NIAMS) grant R01 AR068132, NASA grant 80NSSC18K1473 and Alice T. and William H. Goodwin, Jr. Research Endowment.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
DeNapoli, R.C., Buettmann, E.G., Donahue, H.J. (2022). Cellular and Molecular Biology in Bone Remodeling. In: Takahashi, H.E., Burr, D.B., Yamamoto, N. (eds) Osteoporotic Fracture and Systemic Skeletal Disorders. Springer, Singapore. https://doi.org/10.1007/978-981-16-5613-2_1
Download citation
DOI: https://doi.org/10.1007/978-981-16-5613-2_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-5612-5
Online ISBN: 978-981-16-5613-2
eBook Packages: MedicineMedicine (R0)