
Abstract
New copper(II) complexes—dimeric-[Cu(nphen)(gly)(H2O)]+ (1) and [Cu(dmphen)(gly)(NO3)(H2O)] (2) (nphen = 5-nitro-1,10-phenanthroline, dmphen = 4,7-dimethyl-1,10-phenanthroline, and gly = glycine)—have been synthesized and characterized by CHN analysis, single-crystal X-ray diffraction techniques, FTIR, EPR spectroscopy, and cyclic voltammetry. The CT-DNA-binding properties of these complexes have been investigated by thermal denaturation measurements and both absorption and emission spectroscopy. The DNA cleavage activity of these complexes has been studied on supercoiled pUC19 plasmid DNA by gel electrophoresis experiments in the absence and presence of H2O2. Furthermore, the interaction of these complexes with bovine serum albumin (BSA) has been investigated using absorption and emission spectroscopy. The thermodynamic parameters, free-energy change (ΔG), enthalpy change (ΔH), and entropy change (ΔS) for BSA + complexes 1 and 2 systems have been calculated by the van’t Hoff equation at three different temperatures (293.2, 303.2, and 310.2 K). The distance between the BSA and these complexes has been determined using fluorescence resonance energy transfer (FRET). Conformational changes of BSA have been observed using the synchronous fluorescence technique. In addition, in vitro cytotoxicities of these complexes on tumor cell lines (Caco-2, A549, and MCF-7) and healthy cells (BEAS-2B) have been examined. The antimicrobial activity of the complexes has also been tested on certain bacteria cells. The effect of mono and dimeric in the above complexes is presented and discussed.
Graphical Abstract
New copper(II) complexes—dimeric-[Cu(nphen)(gly)(H2O)]+ (1) and [Cu(dmphen)(gly) (NO3)(H2O)] (2) (nphen = 5-nitro-1,10-phenanthroline, dmphen = 4,7-dimethyl-1,10-phenanthroline and gly = glycine)—have been synthesized and characterized by CHN analysis, single-crystal X-ray diffraction techniques, FTIR and EPR spectroscopy. They have been tested for their in vitro DNA/BSA interactions by the spectroscopic methods. These complexes exhibited higher cytotoxic and antimicrobial activities. Complex 1 shows better DNA / BSA interactions in comparison to complex 2.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Inorganic compounds have been used in medicine for many centuries, though often only in an empirical way with little attempt to design the compounds to be used, and with little or no understanding of the molecular basis of their mechanism of action [1]. In the late 1960s, the discovery and development of the antitumor compound cisplatin played a profound role in establishing the field of medicinal inorganic chemistry [2]. Platinum-based complexes had been a primary focus of the research on chemotherapy agents, but these complexes show severe side effects and acquired resistance [3–5]. The interests in this field have recently shifted to non-platinum-based agents to find different metal complexes [6–8]. Copper complexes could be good candidates owing to the fact that copper has been recognized as a bio-essential element for humans since 1928 [9, 10], and the characterization of its role for biological applications and potential pharmacological activity has been extensively addressed in the development of copper-based drugs [11–14]. Copper is a bio-essential transition metal ion. Its complexes with tunable coordination geometries in a redox-active environment could find better applications at the cellular level. This makes it practical to design tunable transition metal complexes as anticancer drugs to be the most promising alternatives to cisplatin [15]. Casiopeínas® are among the most representative copper complexes with an antitumor potential. The general formula of Casiopeínas® is [Cu(N–N)(L–L’)H2O]NO3 (where N–N diimines are compounds, such as 1,10-phenanthroline, 2,2-bipyridine, or their substituted derivatives, and L–L′ have N–O donor atoms, such as in the aminocidates, or O–O donor atoms, such as from acetylacetonate and salicylaldehydate). The abbreviations for the Casiopeínas® complexes from I to VIII are given as Casiopeína II-gly or Cas IIgly. These complexes have been patented and registered under the name of Casiopeínas® [16–18] and have been tested in several models in vitro and in vivo, showing antiproliferative [19], cytotoxic [20], genotoxic [21], and antitumor activities [22] with promising results [23]. Preclinical studies have revealed that the substitution on the diimine ligand as well as changes in the co-ligand modify the magnitude of the biological activity [24].
Substituted 1,10-phenanthrolines are nitrogen-donor bidentate ligands with a relatively high affinity for copper. Their extended aromatic ring system allows these ligands to bind to DNA by intercalative and non-intercalative interactions either as free ligands or in metal complexes [25]. The amino acids are the chemical units of proteins that constitute the structure for all living organisms [26] and are essential for various biochemical processes that support the maintenance of life in individual organisms [27]. They are good chelating agents [28] and can coordinate with transition metals either through their amino or carboxyl groups [29].
In addition to DNA, protein is also considered to be one of the molecular targets in the action of anticancer agents, which are believed to contribute to unwanted drug side effects, drug resistance, and even possibly drug delivery and storage [30]. Therefore, researchers’ attention has been increasingly directed toward investigating the interactions between drugs and protein, particularly serum protein. Thus, we have investigated the protein-binding ability of the complexes using a serum protein, such as bovine serum albumin (BSA).
Quite recently, we reported the synthesis, characterization, DNA interactions, and cytotoxic activities of some binary and ternary copper(II) complexes with 1,10-phenanthroline or substituted phenanthrolines and tyrosine ligands [31, 32]. Considering the facts mentioned above, in this paper, our group has focused on the synthesis and property studies (CHN analysis, single-crystal X-ray diffraction techniques, FTIR, EPR spectroscopy, and cyclic voltammetry) of mono- and dimeric ternary copper(II) complexes with substituted phenanthrolines (dmphen and nphen) and gly ligands. The detailed structure of two new mono- and dimeric ternary copper(II) complexes with the formulas of [Cu(nphen)(gly)(H2O)]2(NO3)2 (1) and [Cu(dmphen)(gly)(NO3)(H2O)] (2) has also been determined by single-crystal X-ray analysis. Complex 1 is different in structure to Cas Vgly-[Cu(nphen)(gly)(H2O)]NO3 due to dimeric structure, coordination geometry, and dicationic complex. In addition, complex 2 is different in structure to Cas IIgly-[Cu(dmphen)(gly)(H2O)]NO3. Complex 2 is a neutral complex, while Cas IIgly is monocationic complex.
In this study, the biological properties of complexes 1 and 2 were evaluated for (1) their interaction with calf thymus CT-DNA investigated by UV spectroscopy, (2) their ability to displace the classical DNA intercalator ethidium bromide (EB) from its CT-DNA-EB complex as a means to investigate the existence of a potential intercalation monitored by fluorescence emission spectroscopy, (3) thermal denaturation, (4) their interaction with plasmid DNA (pUC19 DNA) studied by agarose gel electrophoresis, (5) their affinity to bovine (BSA) serum albumin investigated by fluorescence spectroscopy, (6) their cytotoxic activity as identified against certain cell lines, and (7) their antimicrobial activity by determining the inhibitory concentration against certain bacteria cells.
Materials and methods
All chemicals were of reagent grade, purchased from different sources, and were used without further purification. 4,7-dimethyl-1,10-phenanthroline and 5-nitro-1,10-phenanthroline were purchased from Alfa-Aesar. Copper(II) nitrate trihydrate, copper(II) chloride, glycine, methanol, KOH, NaCl, tris-(hydroxymethyl)aminomethane-HCl, CT-DNA, BSA, agarose (molecular biology grade), ethidium bromide, bromophenol blue, xylene cyanol, agar, RPMI-1640, fetal calf serum, PBS, and penicillin–streptomycin were provided from Sigma-Aldrich. Tris Borate–EDTA buffer solution (TBE, 10×) was purchased from Sigma. XTT cell proliferation assay was obtained from Biological Industries, Israel. H2O2 (35%, Darmstadt Germany) was used as positive control at a single concentration of 100 μM. In cytotoxicity studies, sterile distilled water was used as solvent control at a maximum concentration of 0.5% (v/v). Cisplatin was purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA. The pUC19 DNA was obtained from Biolabs, UK. All the bacterial strains were obtained from Uludag University, Faculty of Medicine, Department of Medical Microbiology, Bursa, Turkey. All solutions were prepared with analytical grade water (R = 18 MΩ) using grade A glassware. C, H, and N elemental analyses were performed using a Costech elemental analyzer at the Technical and Scientific Research Council of Turkey, TUBITAK Bursa Test, and Analysis Laboratory. Infrared spectra were recorded in the 4000–400 cm−1 region with a Thermo-Nicolet 6700 Fourier-Transform Infrared Spectrometer using KBr pellets. The EPR powder spectrum was recorded on a Bruker EMX X-band spectrometer (9.8 GHz), with about 20 mW microwave power and 100 kHz magnetic field modulation. The magnetization measurements in the temperature range of 10–300 K were carried out on a Quantum Design PPMS system, under the constant magnetic field of 1kOe. The ESI mass spectra were recorded using a Bruker Daltonics Microtof II-ESI-TOF mass spectrometer. Electrochemical behaviors were investigated using a CH Instrument 440B electrochemical analyzer. The spectrophotometric studies were performed using a GBC Cintra 303 UV–Visible spectrophotometer connected with a Peltier thermocell. Emission intensity measurements were carried out using a Jasco FP-750 spectrofluorometer. The DNA bands were visualized under UV light, and photographs were taken. Absorbance measurements in cytotoxicities studies were read using ELx800 Microplate Reader (Bio-Tek, Winooski, VT, USA).
Synthesis of [Cu(nphen)(gly)(H2O)]2(NO3)2 (1)
Complex 1 was prepared by a general synthetic method in which a mixture of nphen (0.5 mmol) and copper(II) nitrate trihydrate (0.5 mmol) in 20 mL methanol was added dropwise to an aqueous solution (5 mL) of glycine (0.5 mmol) and KOH (0.5 mmol) with stirring for about 20 min. The resulting solution was left to evaporate slowly at room temperature. After 6 days, the blue crystals were obtained. Although we did not design dimeric complex formation, dimeric complex was formed at the end of the crystallization. Complex 1. Yield was 80%. Melting point: 286 °C. Anal. calcd. for C28 H26 Cu2 N10 O16 (885.67 g mol−1) (%): C, 37.97; H, 2.96; N, 15.82. Found: C, 38.04; H, 3.02; N, 15.94. ESI–MS (m/z): 362 for [Cu(nphen)(gly)]+. IR (KBr phase, cm−1; w, weak; m, medium, s, strong): ν(C=N)nphen 851 m, ν(C=C)nphen 720 m, ν(COOasym)gly 1602 s, ν(COOsym)gly 1254 m, ν(M–N)nphen 536w, ν(M–O)gly 429w, (NO3 −) 1392 m. UV–Vis [λ nm; Tris–HCl buffer (5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2)]: 274, 608. EPR g ⊥ = 2.079, g // = 2.231.
Synthesis of [Cu(dmphen)(gly)(NO3)(H2O)] (2)
Dark green crystals suitable for X-ray single-crystal analysis were obtained by the same method of preparing complex 1 except using dmphen instead of nphen. Complex 2. Yield was 84%. Melting point: 224 °C. Anal. Calcd. for C16H20CuN4O7 (443.90 g mol−1) (%): C, 43.29; H, 4. 54; N, 12.62. Found: C, 43.33; H, 4.61; N, 12.69. ESI–MS (m/z): 345 for [Cu(dmphen)(gly)]+. IR (KBr phase, cm−1; w, weak; m, medium, s, strong): ν(C=N)nphen 857 m, ν(C=C)nphen 731 m, ν(COOasym)gly 1592 s, ν(COOsym)gly 1264 m, ν(M–N)nphen 532w, ν(M–O)gly 434w, (NO3 −) 1386 m. UV–Vis [λ nm; Tris–HCl buffer (5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2)]: 273, 672. EPR g ⊥ = 2.067, g // = 2.267, a // = 91 G and a ⊥ = 7.6.
X-ray crystallography
The solid-state structures of complexes 1 and 2 were confirmed by X-ray diffraction analysis. Data for complexes 1 and 2 were obtained with Bruker APEX II QUAZAR three-circle diffractometer. Indexing was performed using APEX2 [33]. Data integration and reduction were carried out with SAINT [34]. Absorption correction was performed by multi-scan method implemented in SADABS [35]. Data integration, reductions, and absorption corrections were performed using the SAINT and SADABS programs. The Bruker SHELXTL [36] software package was used for structure solution and structure refinement. All non-hydrogen atoms were refined anisotropically using all reflections with I > 2σ(I). Aromatic and aliphatic C-bound H atoms were positioned geometrically and refined using a riding mode. H atoms of water molecules were located in a difference Fourier map and the O–H distances restrained to be 0.84 Å from O atom using DFIX command and their positions were constrained to refine on their parent O atoms with Uiso(H) = 1.5Ueq(O). The positions of the N-bound H atoms were located from the difference Fourier map and restrained to be 0.89 Å from N atom using DFIX command, and their position was constrained to refine on their parent N atoms with Uiso(H) = 1.2Ueq(N). Crystallographic data and refinement details of the data collection for 1 and 2 are given in Table 1. The selected bond lengths and bond angles are given in Table 2. Crystal structure validations and geometrical calculations were performed using the Platon software [37]. Mercury software [38] was used for visualization of the cif files. Additional crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, and CCDC reference numbers are 1,033,343 for complex 1 and 1,033,342 for complex 2. The data can be obtained available free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 (0)1223336033; email: deposit@ccdc.cam.ac.uk.
DNA interaction studies
The absorption spectral experiments were performed in Tris–HCl buffer (5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2). Solutions of the CT-DNA in Tris–HCl buffer gave the ratio of UV absorbance at 260 and 280 nm, A260/A280, of ca. 1.9, indicating that the DNA was sufficiently free of protein. The concentration of the prepared CT-DNA stock solution was determined according to its absorbance at 260 nm. The molar absorption coefficient, ε 260, was taken as 6600 L mol−1 cm−1 [39]. Stock solution of CT-DNA was stored at 277.2 K and used after no more than 4 days. Concentrated stock solution of complexes 1 and 2 was dissolved Tris–HCl buffer to required concentrations for all the experiments. Absorption spectral titration experiments were performed by keeping the concentration of complexes 1 and 2 constant while varying CT-DNA concentration. After each addition of CT-DNA to the Cu(II) complex, the resulting solution was allowed to equilibrate at 298.2 K for 2 min followed by recording of absorption patterns.
Complexes 1 and 2 did not show luminescence behavior at room temperature in aqueous solution, in any organic solvent examined, or in the presence of CT-DNA. Therefore, the binding of the complexes cannot be directly presented in the emission spectra. Therefore, the fluorescence spectral method using EB as a reference was used to determine the relative DNA-binding properties of complexes 1 and 2 to CT-DNA in Tris–HCl buffer (5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2). For fluorescence experiments, DNA was pretreated with ethidium bromide (EB). Complexes 1 and 2 were then added to this mixture, and their effect on the emission intensity at about 595–605 nm was measured.
DNA thermal denaturation experiments were carried out in Tris–HCl buffer solution (5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2) using CT-DNA (150 μM) by varying the temperature from 293.2 to 373.2 K, both in the absence and presence of complexes 1 and 2 (30 μM). These studies were performed using a GBC Cintra 303 UV–visible spectrophotometer connected with a Peltier thermocell.
The efficiency of DNA cleavage was measured by determining the ability of the complexes to form open circular (OC) and nicked circular (NC) DNA from its supercoiled (SC) form using agarose gel electrophoresis. In reactions using super-coiled pUC19 plasmid DNA, Tris–HCl buffer (50 mM) was diluted with 50 mM NaCl (pH 7.2). The pUC19 plasmid DNA was treated with different dose complexes—(0.5, 1, 12, 25, 50, 100, 200, 400, 800, 1600, 3200, and 6400 μM)—and diluted with the Tris–HCl buffer. The reaction was also carried out in the presence of 5 μM H2O2. Samples were incubated for 4 h at 310.2 K. After incubation, the gel-loading buffer was added, and electrophoresis was performed at 40 V for 1 h in diluted Tris-boric acid-EDTA (diluted TBE, 1×) buffer using 1% agarose gel containing 1.0 μg/mL ethidium bromide. The gel was visualized by photographing the fluorescence of the intercalated ethidium bromide under a UV illuminator. The cleavage efficiency was measured by determining the ability of the complex to convert supercoiled (SC) DNA to its nicked circular (NC) and linear circular (LC) forms.
BSA interaction studies
All experiments involving BSA were performed in Tris–HCl buffer solution (5 mM Tris–HCl/50 mM NaCl buffer at pH 7.2). Final solutions of BSA and complexes 1 and 2 were prepared by dissolving them in the Tris–HCl buffer solution to required concentrations, respectively. The UV–Vis absorption spectra of albumins and the solution of complexes 1 and 2 at 14.75 μM concentration were measured in the range of 200–400 nm. In the tryptophan fluorescence quenching experiment, quenching of the tryptophan residues of BSA was done by keeping the constant concentration of BSA while varying complexes 1 and 2 concentration. The fluorescence measurements were performed at three temperatures (293.2, 303.2, and 310.2 K). The fluorescence spectra were recorded at an excitation wavelength of 280 nm and an emission wavelength of tryptophan residues of BSA at 342 nm after each addition of the quencher. The range of synchronous scanning was λ ex = 280, λ em = 298, and λ em = 342 nm, where the differences in the wavelengths (Δλ) were 15 and 60 nm. BSA was done by keeping the constant concentration while varying complexes 1 and 2 concentration. The three-dimensional fluorescence scanning was λ ex = 230/280.
Cytotoxicity assay (XTT)
The cytotoxicities of complexes 1 and 2 and cisplatin were evaluated against three cancer cell lines, including A549, MCF-7, Caco-2, and one healthy cell line BEAS-2B (kindly provided from Feray KOCKAR) using the XTT assay. Cells were cultured in RPMI-1640 medium supplemented with 15% fetal calf serum (FCS), penicillin (100 IU/mL) and streptomycin (100 μg/mL), 10 mM l-glutamine, 10 mM non-essential amino acids, and sodium pyruvate. Cells were maintained at 310.2 K in a humidified atmosphere containing 5% CO2. Cells were grown in 75 cm2 flasks and subcultured once a week. Growing cells were harvested and counted, and A549 and BEAS-2B cells were seeded 5 × 103 cells/well and Caco-2 and MCF-7 cells were seeded 8 × 103 cells/well in sterile 96-well flat-bottom plates in triplicate. The cells were incubated overnight at 310.2 K in a humidified atmosphere containing 5% CO2. The plates include blanks which containing complete medium without cells. The copper(II) complexes were dissolved in sterile distilled water. Cells were then treated with various concentrations of copper(II) complexes for 24 h. Under the same experimental conditions, cisplatin was also evaluated as a reference metallodrug. The cytotoxicity was measured using XTT Cell Viability Kit. XTT, a tetrazolium salt, is reduced by metabolic active cells to orange formazan dye. After treatment for 24 h, wells were washed with PBS and the medium was replaced with fresh medium (100 μL). The activated XTT solution (50 μL) was added to each well according to the manufacturer’s instructions (Biological Industries). The plate was incubated for additional 3 h in the CO2 incubator at 310.2 K. Absorbance was read using Microplate Reader at a wavelength of 450 nm, and a cell proliferation curve was drawn. After blank subtraction, percent growth inhibition of cells was calculated as follows:
Antimicrobial studies
The antibacterial activities of synthesized complexes were evaluated against Escherichia coli (ATCC 35218), Escherichia coli (ATCC 25922), Enterococcus faecalis (ATCC 29212), Salmonella typhimurium (ATCC 14028), Staphylococcus aureus (ATCC 25923), Yersinia enterocolitica (ATCC 9610), and Klebsiellapneumoniae (ATCC 700603). Cu(II), complexes 1 and 2 were prepared in aqueous solution, while nphen and dmphen were prepared in ethanol. The antibacterial activities of all solutions were determined by agar well diffusion method. The cell concentration of all test microorganisms was adjusted at 0.5 McFarland turbidity standards, and inoculated on nutrient agar plates using sterilized cotton swabs. Wells were made in the inoculated agar medium under sterile conditions (7 mm in diameter), and 100 mL of samples in different concentrations (12.5–400 µg/mL) was pipette into the wells. The agar plates were incubated at 310.2 K for 24 h. After this period, the diameters of the inhibition zones formed around each well were determined, and measured in mm. Each test was carried out in triplicate.
Results and discussion
Description of the crystal structures
The asymmetric unit of complex 1 is composed of one Cu(II) ion, one nphen, one gly, one coordinated water molecule, and one uncoordinated nitrate anion, as shown in Fig. 1. At first glance, the crystal structure of complex 1 appears to be a dinuclear complex that contains two [Cu(nphen)(gly)(H2O)]+ units through the weak coordination [Cu1-O3i = 2.745(2) Å] of the carboxylate oxygen (O3) of gly with the central Cu ion. In the [Cu(nphen)(gly)(H2O)]+ unit, the central Cu(II) ion is five-coordinated through the carboxylate oxygen (O2), the amino nitrogen (N3) atom of gly, the two N-donor atoms of one chelating nphen ligand, and one O atom (O1) of the coordinated water molecule, neglecting the weak interaction between the Cu1 and O3 atoms (Cu1–O3i, i: −x, −y, 1 − z). The tau-descriptor (τ 5) value of 0.08 indicates no significant distortion of the geometry (τ 5 = 0 for an ideal square pyramid and τ 5 = 1 for an ideal trigonal bipyramid) [40]. The shortest Cu–Cu distance is 5.076 Å. In the CuN3O2 coordination sphere, equatorial bond lengths [(Cu1–N1 = 2.018(2) Å, Cu1–N2 = 2.002(2) Å, Cu1–N3 = 1.979(2) Å, and Cu1–O2 = 1.9559(17) Å] and the weakly bound axial bond length [Cu1–O1 = 2.291(2) Å] fall in the normal range, and agree well with those reported for the Cu(II)-glycinato complex [41]. The N–Cu–N, N–Cu–O, and O–Cu–O bond angles range from 81.78(8)° to 176.92(9)°. The crystal structure of complex 2 is shown in Fig. 2. The asymmetric unit consists of one Cu(II) ion, one dmphen, one gly, one coordinated nitrate anion, and one coordinated water molecule. The Cu(II) center displays an octahedrally elongated geometry by the two N-donor atoms of one chelating dmphen ligand, the carboxylate oxygen (O2) and the amino nitrogen (N3) atoms of glycine, and the two O donors (O1 and O6) of the aqua and the nitrate ligands. In the CuN3O3 coordination sphere, the equatorial bond lengths [Cu1–N1 = 1.9884(16) Å, Cu1–N2 = 2.0283(16) Å, Cu1–N3 = 1.9993(17) Å, and Cu1–O2 = 1.9550(13) Å], and weakly bound axial bond lengths [Cu1–O1 = 2.3875(16) Å and Cu1–O6 = 2.5724(16) Å] agree well with those reported for the corresponding Cu(II) complex [42]. The N–Cu–N, N–Cu–O, and O–Cu–O bond angles range from 81.66(6)° to 176.21(7)°.

Crystal structure of complex 1. The uncoordinated nitrate anion is omitted for clarity. Symmetry code: (i): −x, −y, and 1 − z

Crystal structure of complex 2. The lattice water molecule is omitted for clarity
Spectral characterization
The IR spectra showed peaks associated with ligands coordinated to a copper(II) metal ion. Compared to the free ligands, ν(C=N) and ν(C=C) of the ligands were found to change slightly in the IR spectra of the complexes. The IR spectra of complexes 1and 2 displayed peaks at 851 and 720 cm−1 for complex 1 and 857 and 731 cm−1 for complex 2. These peaks were assigned as coordinated ν dmphen and ν nphen skeletal vibrations. The IR spectra of complexes 1 and 2 demonstrated the presence of water molecules, as indicated by a broad band of ν(O–H) at 3403–3481 cm−1, which is in accordance with the results of the elemental analyses. Two medium-to-weak peaks appear in the region of ~3300 cm−1 for the N–H stretching of the aminoacidato groups. The strong peaks appearing in the ~1600 cm−1 region were assigned to ν(COO) but suffered interference from the ν(C=N) and δ(NH) peaks. Gly is coordinated to the metal ion as an N,O-bidentate ligand, which is in agreement with the result obtained by single-crystal X-ray diffraction for complexes 1 and 2. The very strong broad peaks in the ~1390 cm−1 region suggest discrete nitrate anions, in agreement with the X-ray structure of complexes 1 and 2.
To obtain further structural information, the electronic spectra of complexes 1 and 2 were measured in the UV–Vis region (200–700 nm). The electronic bands appeared in the UV region in the wavelength range of 220–290 nm correspond to π–π* intra-ligand transitions associated with the coordinated ligands in complexes 1 and 2. Both of the electronic spectra exhibited a very broad and weak absorption band in the visible region at 608 and 672 nm which is attributed to the d–d ligand-field transitions of the central copper(II) ions. The complexes showed good solubility in water, ethanol, DMF and DMSO, and insolubility in hydrocarbons. They were found to be stable in both the solid and solution phases.
The values of molar magnetic susceptibility of complexes 1 and 2 were recorded in the temperature range of 10–300 K, and the plots of the molar magnetic susceptibility (χ m) and χ m T for these complexes are shown in Fig. 3a and b. The variable temperature dependences of χ m for complexes 1 and 2 were fitted by the relation of \(+ C/T,\) where C is the Curie constant and α is temperature independent susceptibility (TIP) [43]. From this fitting process, \(C = 0.40000 \pm 0.00005\;{\text{emuK/mol}} . {\text{Oe}}\), \(\alpha = 0\) for complex 1; \(C = 0.42848 \pm \frac{{0.00008\;{\text{emuK}}}}{\text{mol}} . {\text{Oe}},\) and \(\alpha = 0.00014 \pm \frac{{0.0000004\;{\text{emu}}}}{\text{mol}} . {\text{Oe}}\) for complex 2 were determined. The TIP arises from the orbital moments of d electrons and Pauli paramagnetism. The effective magnetic moment, \(\mu_{\text{eff}}\), was calculated to be 1.79 for complex 1 and 1.85 for complex 2 using the relation \(\mu_{\text{eff}} = 2.83(C)^{1/2}\) in Bohr magneton (\(\mu_{\text{B}}\)). The values of \(\mu_{\text{eff}}\) seem above the spin only value due to mixing of some spin–orbital angular momentum from excited states via spin-orbit coupling [44]. From these results, we conclude that these complexes only show a paramagnetic behavior, that is, there are no, magnetically, intra- and inter-molecular interactions in these structures.

Temperature dependence of the molar magnetic susceptibility χ m and χ m T for a complex 1 and b complex 2. Solid line represents a fit by the Curie law
Figure 4a shows the powder EPR spectrum of complex 1 at the room temperature. The spectrum displays two groups of lines overlapping lines, having strong and weak intensities, indicating an axial symmetry around the Cu(II) ion (S = 1/2, I = 3/2) in the complex. The hyperfine structure could not be resolved due to line broadening caused by spin-orbital and spin-exchange interactions because of the excess spin concentration. The spectrum was simulated using the simFonia computer program. The best simulating parameters are determined to be g ⊥ = 2.079 and g // = 2.231. The order of g // > g ⊥ > g e (free-electron g value, g e = 2.0023) shows that the Cu(II) ions are located in tetragonally distorted octahedral sites (D 4h) elongated along the z-axis and the ground state of the paramagnetic electron is \(d_{{x^{2} - y^{2} }}\) (2 B 1g state). The value of less than 2.3 of g z (g //) shows the covalent character of the metal–ligand bonding in this complex. Figure 4b shows the powder EPR spectrum of complex 2 at room temperature. In the spectrum, two groups of lines are seen overlapping and have strong and weak intensities, thus indicating an axial symmetry around the Cu (II) ion (S = 1/2, I = 3/2) in the complex. The hyperfine structure could partly, but not entirely, be seen due to line broadening caused by spin-orbital and spin-exchange interactions because of the excess spin concentration. The spectrum was simulated with the values of g, a (hyperfine coupling constant) and linewidth extracted from the powder spectrum, using the simFonia computer program. The best simulating parameters are determined to be g ⊥ = 2.067, g // = 2.267, a // = 91 G, and a ⊥ = 7.6. The order of g // > g ⊥ > g e (free-electron g value, g e = 2.0023) shows that Cu(II) ions are located in tetragonally distorted octahedral sites (D 4h) elongated along the z-axis, and the ground state of the paramagnetic electron is \(d_{{x^{2} - y^{2} }}\) (2 B 1g state) [45–48]. The free-electron value of g is changed by the orbital effect. However, the covalent character of the metal–ligand bond reduces this effect. Thus, this reduction also decreases the amount of deviation of the g value from that of the free electron [44]. According to this, the value of less than 2.3 for g z (g //) shows the covalent character of the metal–ligand bonding in this complex [49].

Powder EPR spectrum of complexes at room temperature and its simulation. a Complex 1. b Complex 2
The cyclic voltammetric studies of complexes 1 and 2 were studied in DMSO solution containing TBAP as supporting electrolyte at room temperature. The current–potential curves for 10−3 M solutions of complexes 1 and 2 using 0.1 M TBAP in DMSO are demonstrated in Fig. 5a and b. The first redox couples −0.13/−0.08 V in complex 1 and 0.05/−0.1 V in complex 2 are reversible. This wave may be assigned to CuII/CuI redox couple. Complexes 1 and 2 show another oxidative wave at +0.19 V as well. This wave is irreversible. This suggests that the metal-centred species corresponding to this wave is again unstable in solution and reverts back to the original species. Hence, this irreversible oxidation wave may be assigned to the oxidative couple CuIII/CuII.

CV of a complex 1 and b complex 2 on a glassy carbon working electrode, Ag/Ag+ reference electrode, and Pt counter electrode in DMSO, with a complex concentration of ca. 10−3 M, 0.1 M TBAP as supporting electrolyte, and a scan rate of 100 mV s−1
To determine active chemical species of the complexes, electrospray ionization mass spectrometry (ESI–MS) analysis was performed. The mass-to-charge ratio peaks observed at m/z 362 and 345 were due to the formation of the species [Cu(nphen)(gly)]+ for complex 1 and [Cu(dmphen)(gly)]+ for complex 2, respectively. The coordinated water molecule dissociates under the ESI–MS conditions, as observed in ESI–MS of other copper(II) complexes [50, 51]. Although complex 2 was a neutral in a solid state, mass spectral peaks show that correspond to the species showing loss of nitrate and water ligand, so complex 2 can be dissolved in water.
DNA binding and cleavage activities
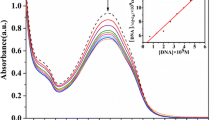
Electronic absorption spectroscopy is an effective method of examining the binding mode and binding extent of metal complexes with DNA. Hypochromism and red shift are generally associated with the intercalation of the metal complexes to the DNA helix, because the intercalative mode involves a strong stacking interaction between the aromatic chromophore of the complexes and the base pairs of the DNA [52]. The interaction of complexes 1 and 2 with CT–DNA has been studied with UV spectroscopy to investigate the possible binding modes to CT–DNA and calculate the binding constants (K b) to CT–DNA. The results of the absorption spectra of complexes 1 and 2 in the absence and presence of CT-DNA are shown in Fig. 6. Upon increasing the ratio of the concentration of DNA to those of complexes 1 and 2, the absorption bands of complexes 1 and 2 exhibited hypochromism of 10 and 42%, with blue shifts of 1 and 1.3 at 274 and 273 nm, respectively. From the absorption data, a plot was drawn of [DNA]/(ε a–ε f) values versus DNA concentrations for the titration of DNA to complexes 1 and 2 using the following equation [53]:
where [DNA] is the concentration of DNA in terms of base pairs, ε a is the apparent extinction coefficient obtained by calculating A obs/[complex], ε f corresponds to the extinction coefficient of the complex in its free form, and ε b refers to the extinction coefficient of the complex in the bound form. Each set of data, when fitted to the above equation, gave a straight line with a slope of 1/(ε b – ε f) and a y-intercept of 1/K b(ε b – ε f). Finally, the K b values were calculated (Fig. 6). The logarithms of the K b values (log K b) for complexes 1 and 2 were 3.86 and 3.83, respectively, which follow the order 1 > 2. These outcomes are listed in Table 3.

UV–Vis absorption spectra of complexes 1 and 2 upon the addition of CT-DNA ([CT-DNA] = 14.78 μM; [Complexes] = 0–500 μM). Inset plot of [DNA]/(ε a − ε f) versus [DNA] for the titration of DNA to complexes
Based on our previously studies, we found that DNA interactions and cytotoxic activities of binary and ternary copper(II) complexes are higher than those of the corresponding free ligands [31, 32]. Despite different copper(II) complexes, various ligands, and coordination environments, the DNA/BSA interactions of complexes 1 and 2 have been compared with those previously reported complexes [54–58]. The literature has already shown that phenanthroline derivatives and amino acids are good DNA and protein binders which in the present study have been found to be further enhanced on complexation with copper(II). This could be based on their better binding constants obtained for interactions with DNA and BSA compared to those of the free ligands [59]. In addition, a much less or no DNA cleavage activity was exhibited by the ligands when compared to that of Cu(II) complexes [60–62]. This could be explained by the fact that two possible reasons. (1) The electrostatics of positively charged Cu(II) ions cause an interaction with polyanionic DNA. (2) The charge transfer of coordinated ligand, caused by coordination of the central Cu(II), results in reduction of charge density of the planar conjugated system; this change will lead to complexes binding to DNA more easily [63–65]. Therefore, in this study, we did not discuss the less effect of the ligands.
The binding constant of complex 1 was observed to be comparatively larger than that of complex 2. The calculated binding constant (K b) of these complexes to CT-DNA was found to be of lower magnitude than that of the classical intercalator EB (ethidium bromide) (K b = 1.23 × 105 M−1) [66]. However, the K b value for these complexes is comparable to that observed for other copper(II) complexes, such as [Cu(2,2′-bipyridine)(glycine)Cl]2H2O (K b = 1.84x103) and [Cu(2,2′-dipyridylamine)(glycine)Cl]2H2O (K b = 3.1 × 103) [67], [Cu(1,10-phenanthroline) (glycine)(H2O)]NO3·1.5H2O (K b = 4.68 × 103) [55], [Cu(2-(2′-pyridyl)benzothiazole) (glycine)(H2O)]ClO4 (K b = 7.16 × 103) [68], [Cu(glycyl-glycine)(2-(4′-thiazolyl)benzimidazole) (Cl)]4H2O (K b = 1.64 × 105) [69], and [Cu(dipyrido[3,2-a:2′,3′-c]phenazine)(glycine)]ClO4 (K b = 2.10 × 106) [70]. From our absorption spectroscopy results, we can suggest that a moderate intercalative binding mode of complexes 1 and 2 with CT-DNA is proposed based on the data.
The absorption titration results indicate that the complexes effectively bind to DNA. To confirm the binding mode and compare their binding affinities, EB displacement experiments were carried out. The EB-DNA system generally shows a significant increase in the fluorescence emission when EB binds to DNA, and a decrease is observed when EB is displaced by another DNA intercalate molecule [71]. The results of fluorescence titration spectra have also been confirmed to be effective for characterizing the binding mode of the metal complexes to DNA. The fluorescent emission titration of complexes 1 and 2 is illustrated in Fig. 7. An increase in the concentration of the complex results in a decrease in the emission intensity of the EB-DNA solution. The apparent binding constant (K app) [72] is calculated using Eq. 3 and is the concentration of the compound in the medium at a 50% reduction of the fluorescence intensity of the EB. The logarithms of the K app values were calculated to be 6.00 and 5.36, respectively, for complexes 1 and 2. With the addition of the compound to CT-DNA, there was a noticeable decrease in the emission intensity due to the replacement of EB by the complexes.
where [Complex] is the concentration of the ternary complex at 50% reduction of fluorescence intensity (K EB = 1.0 × 107 M−1 and [EB] = 1.3 μM) [73]. The quenching plots followed the Stern–Volmer relationship of the form [74]:
where I 0 and I are the fluorescence intensities of the excited CT-DNA + EB in the absence and presence of the complexes. K sv is the Stern–Volmer quenching constant and [Complex] is the concentration of the quencher. From the experimental data, a plot of the emission intensity (I/I 0) versus compound concentration was prepared (Fig. 7). These behaviors were analyzed with Eq. 4, and the log K sv values were calculated to be 4.36 and 3.82, respectively, for complexes 1 and 2. In addition, the Stern–Volmer quenching constants (K sv) and K app are shown in Table 3.

Effect of the addition of complexes 1 and 2 on the emission intensity of the CT-DNA bound EB at different concentrations ([EB] = [CT-DNA] = 5 μM; [Complex] = 0–200 μM, r = [Complex]/[DNA]), Inset Stern–Volmer plot of the fluorescence data
Comparison of EB displacement properties between complexes 1 and 2 and previously reported compounds revealed that these complexes have comparable binding ability with those of previously reported complexes [55, 67–70]. However, the K SV value of complexes 1 and 2 is lower than that observed for classical intercalator EB (107 M−1) [75] which imply that these complexes bind with DNA relatively less strongly than classical intercalators. This could be explained by the fact that some aromatic planar ligands show moderate binding strength [76].
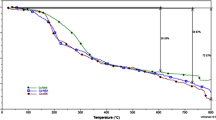
DNA denaturation, commonly known as DNA melting, is the process by which double-stranded DNA unwinds and separates into single strands through the breaking of the hydrogen bonding between the bases. Intercalation of natural or synthesized organics and metallointercalators generally results in a considerable increase in the melting temperature (T m) [77]. Classical intercalators, such as EB, are known to show significantly high ΔT m values (11.2 °C) [78]. In this experimental medium, EB displays remarkable ΔT m values of 11.8 °C. The DNA denaturation experiment shows a significant shift in the melting temperature (T m), yielding a ΔT m value of 4.6–8.9 °C upon the addition of the compounds to CT-DNA. The effects on the melting curves show clearly that these complexes bind to DNA. The results are concordant with the absorption and fluorescence conclusions.
The DNA cleavage experiment of complexes 1 and 2 was investigated using supercoiled pUC19 plasmid DNA by electrophoresis in the absence and presence of H2O2. When the original super-coiled form (Form I) of plasmid DNA is nicked, an open circular relaxed form (Form II) will exist in the system and the linear form (Form III) can be found upon further cleavage. During electrophoresis, the compact Form I migrates relatively faster, while the nicked Form II migrates more slowly, and the linearized form (Form III) migrates at rate between those of Forms I and II. Figure 8a, b shows the electrophoresis results of the DNA cleavage induced by increasing the concentrations of complexes 1 and 2 to 0.5, 1, 12, 25, 50, 100, 200, 400, 800, 1600, 3200, and 6400 μM without H2O2. Each plasmid DNA sample exhibited an obvious variation in its electrophoretic mobility, as indicated in lines 2–13. Control experiments with plasmid DNA (Line 1 in Figs. 8a, b, 9a, b) did not show any apparent conversion of Form I to its Form II. With an increasing complex concentration, the amount of Form I decreased gradually with a concomitant increase in Form II. Complexes 1 and 2 displayed different cleavage patterns under the same experimental conditions. When the concentration reached 12 µM, complex 1 promoted a complete conversion from Form I to Form II (Fig. 8a). However, under the same conditions (Fig. 8b), complex 2 showed almost no effect on the cleavage of DNA even at a concentration of 12 µM. At 200 µM, complex 2 shows complete conversion of Form I of the DNA to its Form II. These results demonstrate that both complexes show DNA cleavage activity, but complex 1 shows a higher activity than complex 2. This indicates a possible synergistic effect between the two copper ions in the dimeric complex 1 that contributes to its relatively high nucleolytic efficiency.

Agarose gel electrophoresis diagrams. Cleavage of pUC19 plasmid DNA induced by a complex 1 and b complex 2. Lane 1 control, lanes 2–13 DNA + complex: 0.5, 1, 12, 25, 50, 100, 200, 400, 800, 1600, 3200, and 6400 μM, respectively

Agarose gel electrophoresis diagrams. Cleavage of pUC19 plasmid DNA induced by a complex 1 and b complex 2. Lane 1 control, lanes 2–7 DNA + complex + H2O2: 0.5, 1, 12, 25, 50, and 100 μM, respectively
Figure 7a and b reveals that cleavage of pUC19 DNA is induced by complexes 1 and 2 in the presence of H2O2 at concentrations of 0.5, 1, 12, 25, 50, and 100 μM. In the presence of H2O2, the complexes cleave DNA more effectively which may be due to the reaction of the hydroxyl radical with DNA. With the same concentration (0.5 μM) of complexes 1 and 2, Form I was converted to Form III or was even more cleaved to smaller fragments (Fig. 9a, b, lane 2). These phenomena suggest that complexes 1 and 2 have the highest cleavage efficiency in the presence of H2O2. It is known that H2O2 initiates a series of reactions to form HO˙ radicals. The radicals thus formed attack the DNA strand and cause DNA breakage. The extent of DNA cleavage induced by both complexes that result in the linear form of DNA (Form III) occurs at lower doses with H2O2. It is important here to compare the percentage of DNA cleavage versus the concentration of related binary Cu(II)-phen derivatives and ternary Cu(II)-phen derivatives-amino-acid complexes [31, 32, 55, 60–62, 79] in the absence and presence of external agents (Table 4). On comparison of the cleavage property with some reported results [55, 60–62] of Cu(II) complexes, we pointed out that a lesser amount of the reducing agent and complex are required in our case for maximum cleavage of DNA. This is well reflected from the cleavage data which show that more than 75% of cleavage occurs with a low complex concentration.
Based on a comprehensive literature results, a tentative mechanism is suggested for the hydrolytic pathway. Copper(II) complexes show distorted octahedral and tetrahedral symmetries due to d 9 configuration. Therefore, typical copper(II) complexes have square planar or square pyramidal geometries with weakly associated ligands in the axial position [80]. The distortion is usually seen as axial elongation consistent with the lability and geometric flexibility. Single-crystal X-ray diffraction analysis reveals that complexes 1 and 2 have a weakly bound axial bond length [Cu1–O1 = 2.291(2) Å] and [Cu1–O1 = 2.33875(16) Å], respectively. The high lability of hydrated Cu(II) ion is attributed to the dynamic Jahn–Teller effect [81, 82]. Therefore, dissociation of a water molecule is reasonably easy. It was mentioned earlier that whenever a H2O molecule is coordinated to a metal ion in axial position, this H2O molecule is activated, which produces a nucleophilic M–OH species that eventually participates in DNA cleavage [83–87]. An intramolecular pathway, where an HO˙ is transferred from the metal to a phosphate group of DNA, was also suggested for the hydrolysis of phosphate diesters [88–92]. We can suggest that a similar DNA cleavage activity can be demonstrated complex 1 and 2 which have a weakly bound water ligand.
Mechanisms for oxidative DNA cleavage activity by compounds are generally used in the presence of various quenchers for the reactive oxygen species. The addition of hydroxyl radical scavengers, such as DMSO, catalase, and KI, shows a significant inhibition of the DNA cleavage activity of compounds, indicating the possibility of the involvement of the hydroxyl radical and/or a “copper-oxo” intermediate as the reactive species. It is well known that reactive oxygen species, such as hydroxyl radical (HO˙), cause damage to the sugar and/or base moieties of DNA [93].
Very recently, Ramakrishna and coworkers have found the ternary Cu(II) complex of phen and aliphatic amino acids (l-alanine, l-leucine, and l-proline) to cleave DNA oxidatively [60–62]. As shown in Table 4, these complexes were found to be DNA cleavage active when treated with MPA. When the standard hydroxyl radical scavengers DMSO, KI or catalase was added to the reaction mixture of complex + DNA + MPA, and the DNA cleavage activity of these complexes decreases significantly (Table 4). Therefore, it could be the probably of formation of hydroxyl radical during the redox reaction involving copper(II) center in the presence of MPA. We can suggest that a similar DNA cleavage activity can be demonstrated complexes 1 and 2 which contain dmphen/nphen and aliphatic amino acid (gly).
On the basis of a comprehensive literature observation, the mechanism of oxidative DNA cleavage may be proposed as follows: DNA cleavage is redox-mediated. The complexes would first interact with DNA by intercalation to form a CuII····DNA adduct species, followed by its reduction by the external agent (H2O2) to a CuI····DNA adduct, which then generates HO˙ radicals on reaction with O2. These HO˙ radicals would then attack DNA, causing strand scission (Eq. 5) [94–96]. The following equation is a similar way as proposed by Sigman and coworkers [97]:
BSA-binding activities
Albumin is the most abundant serum protein in the blood and its interaction is of great interest, because non-covalent conjugation of albumin with a number of drugs has been shown to carry their active molecules in the blood [98]. Because bovine serum albumin (BSA) is structurally homologous with human serum albumin, it was chosen as the model protein in our present work. Therefore, the BSA-binding activities of complexes 1 and 2 were studied through electronic absorption studies, fluorescence quenching, determination of the binding constant and number of binding sites, energy transfer between BSA and complexes 1 and 2, synchronous spectral, and three-dimensional fluorescence studies.
Quenching usually occurs either by dynamic or static quenching. Dynamic quenching is a process in which the fluorophore and the quencher come into contact during the transient existence of the excited state. Conversely, static quenching refers to the formation of a fluorophore-quencher complex in the ground state. A simple method to determine the type of quenching is electronic absorption spectroscopy. In general, the metal complexes that bind with BSA may result in hyperchromism or hypochromism. Figure 10 shows the electronic absorption spectra of BSA in the absence and presence of the complexes. The absorption intensity of BSA was enhanced, as the complexes were added, and there was a blue shift of 2.56 nm for complex 1 and 3.84 nm for complex 2. In addition, hyperchromism reached 51% for complex 1 and 35% for complex 2 in the ratio of [BSA]/[complex] = 1. This showed that there exists a static interaction between BSA and the added complexes due to the formation of the ground-state complex of the type BSA complex.

UV–Vis absorption spectra of complexes 1 and 2 upon the addition of BSA
To examine the protein-binding behaviors of complexes 1 and 2, tryptophan emission-quenching experiments were performed. In general, the fluorescence of protein can be contributed to three intrinsic characteristics of the protein, namely, its tryptophan, tyrosine, and phenylalanine residues. The quenching mechanisms are usually classified into dynamic and static quenching, which can be distinguished by their different dependence on temperature and viscosity, or by lifetime measurements [99]. Since higher temperatures result in larger diffusion coefficients, the bimolecular quenching constants are expected to increase with increasing temperature. In contrast, increasing temperature is likely to result in decrease the stability of complexes; thus, the values of the static quenching constants are expected to be smaller [100]. To study the fluorescence quenching mechanism, the fluorescence quenching data at different temperatures (293.2, 303.2, and 310.2 K) were analyzed using classical Stern–Volmer equation (Fig. 9) [101]:
where I 0 and I are the fluorescence intensities in the absence and presence of the complexes, respectively. [Complex], and K q and τ 0 are the total concentration of complexes 1 and 2, the quenching rate constant and the average lifetime of EB-DNA in the absence of the quencher (τ 0 = 10−8 s) [102], respectively. The Stern–Volmer quenching constants log K sv and log K q are illustrated in Table 5. As shown in Table 4, the Stern–Volmer plots were linear and the values of Ksv decreased with increasing temperature (Fig. 11). Clearly, the rate constants of BSA quenching initiated by complexes 1 and 2 are higher than 2.0 × 1010 M−1 s−1, which is the maximum collision quenching constant of various types of quenchers of biomacromolecules [103] and indicate that the binding of complexes 1 and 2 to BSA is responsible for the quenching of its tryptophan fluorescence by the static quenching mechanism. This line of reasoning was also validated by the following UV absorption spectroscopy studies. The modified Stern–Volmer equation was used to evaluate the association constant, log K a, for BSA + complexes 1 and 2 interaction [104]:
where K a is the modified Stern–Volmer association constant, which equals the quotient of an ordinate 1/f a and slope 1/f a K a for the accessible fluorophores, and ƒ a is the fraction of accessible fluorescence. As shown in Table 5, the decreasing trend of log K a with increasing temperature was in accordance with the dependence of log K sv values on temperature, which coincides with a static-type quenching mechanism.

Effects of complexes 1 and 2 on the fluorescence spectra of BSA. Inset Stern–Volmer plots of BSA quenched by complexes 1 and 2 at three temperatures ([BSA] = 1 μM, [Complex] = 0–20 μM, r = [Complex]/[BSA])
Fluorescence intensity data can also be used to obtain the binding constant (K A) and the number of binding sites (n). For the static quenching interaction, the binding constant (K A) and the number of binding sites (n) can be determined according to Eq. 8. When small molecules bind independently to a set of equivalent sites on a macro-molecule, the equilibrium between free and bound molecules is given by the equation [105]:
where in the present case, K A is the binding constant to a site, and n is the number of binding sites per albumin. The logarithms of K A values (log K A) for BSA + complexes 1 and 2 and number of binding sites (n) for complexes 1 and 2 were listed in Table 5. The values of n for both albumins were approximately equal to 1, suggesting that there was one binding site in both BSA for complexes 1 and 2 during their interaction. Hence, complexes 1 and 2 most likely bound to the hydrophobic pocket located in subdomain IIA or IIIA [106]. In addition, the K A values of BSA + complexes 1 and 2 increased with increasing temperature.
In general, the interaction forces between a ligand and protein may include hydrophobic interactions, electrostatic, van der Waals interactions, hydrogen-bonding forces, etc. To identify the nature of the interaction between complexes 1 and 2 and albumins, thermodynamic parameters are calculated according to van’t Hoff equation:
where R is the gas constant, ΔH is the enthalpy change, and ΔS is the entropy change. In addition, the free-energy change (ΔG) can be calculated by the following equation:
A plot of lnK versus 1/T enables the determination of ΔH and ΔS for the binding at different temperatures. According to the data given in Table 6, the values of ΔH for the binding interaction of complexes 1 and 2 to BSA were found to be 114.26 and 321.31 kJ mol−1, and the values of ΔS for the binding interaction of complexes 1 and 2 to BSA were found to be 485.60 and 1170.15 J mol−1 K−1, respectively. A negative value of ΔG indicates the spontaneity of the interaction. The positive values obtained for both ΔH and ΔS indicate that a hydrophobic association is the major binding force and that the interaction is entropy driven process, while hydrogen bonds and van der Waals force are not the dominating factors. In addition to hydrophobic interaction, a possible covalent bonding may also be considered. However, the value of ΔH of complex 2 obtained here is above what would be expected for a covalent bond formation, which should be ≥120 kJ mol−1 [107].
The efficiency of energy transfer in biochemistry can be used to evaluate the distance between the ligands and the fluorophores in the protein [108]. The rate of energy transfer depends on the extent of the overlapping of the donor emission spectrum with the acceptor absorption spectrum, the relative orientation of the donor and acceptor transition dipoles, and the distance between these molecules [109]. The distance from the tryptophan residue (the donor) to the bound drug (the acceptor) in BSA can be calculated according to Forster’s theory. Figure 12a and b depicts the overlap between the fluorescence emission spectrum of BSA and the absorption spectrum of complexes 1 and 2. Fluorescence resonance energy transfer (FRET) is a very useful tool to measure the distance between the donor and acceptor fluorophores. The energy transfer efficiency is obtained using the equation:
where I and I 0 are the luminescence intensities of albumin in the presence and absence of complexes 1 and 2 (1 μM), r is the distance between the acceptor and the donor, and R 0 is the critical distance when the transfer efficiency is 50% and the value is given by equation:

Overlap of fluorescence emission spectrum of BSA and absorption spectrum of the complexes. [BSA] = [Complexes] = 1 μM. a Complex 1. b Complex 2
Here, F(λ) is the luminescence intensity of the fluorescent donor at the wavelength λ and is dimensionless, and ε(λ) is the molar absorption coefficient of the acceptor at λ and its unit is cm−1 M−1 [110]. The efficiency of energy transfer and overlapping integration values was obtained from Eqs. 12 and 13, respectively. To evaluate the Förster’s critical distance using Eq. 12, we have used K 2 = 2/3, N = 1.336, and ϕ = 0.118 for BSA [111]. Using Eqs. 11–13, we have calculated the values of E, r, R 0, and J and shown in Table 6. The value of r less than 8 nm indicated the nonradiative energy transfer between BSA and complexes 1 and 2 [112], and again, the static quenching interaction has occurred between BSA and complexes 1 and 2.
To investigate in detail the structural changes which occurred to BSA upon the addition of new compounds, synchronous fluorescence spectra of BSA were measured before and after the addition of the test compounds. The results provide reasonable information on the molecular microenvironment, particularly in the vicinity of the fluorophore functional groups. According to Miller [113], the difference between the excitation wavelength and emission wavelength (Δλ = λ em − λ ex) indicates the type of chromophore. When Δλ is 15 nm, synchronous fluorescence is characteristic of tyrosine residue, while Δλ of 60 nm is characteristic of tryptophan residues [114].
The maximum emission wavelengths of tryptophan and tyrosine residue in the protein molecule are related to the polarity of their surroundings; changes of the maximum emission wavelengths can reflect changes of protein conformation. The effect of complexes 1 and 2 on BSA synchronous fluorescence spectroscopy with Δλ = 60 nm and Δλ = 15 nm is shown in Fig. 11a–d.
For complexes 1 and 2, as can be seen from Fig. 13a and f, the characteristic emission spectrum of Tyr in BSA has a slight red shift from 300 to 303 nm. The fluorescence intensities of Tyr residues on BSA decreased 84.2/49.5% in the presence of complexes 1 and 2, respectively. As shown in Fig. 11c and d, the maximum emission of Trp has a slight red shift from 339 to 346 nm for BSA. The fluorescence intensities of Trp residues on BSA decreased 88.2/60.8% in the presence of complexes 1 and 2, respectively.

Synchronous fluorescence spectra of BSA with different concentrations of a complex 1 and b complex 2 at Δλ = 15 nm, c complex 1, and d complex 2 at Δλ = 60 nm. The synchronous fluorescence quenching degree of BSA by e complex 1 and f complex 2 at Δλ = 15 nm and Δλ = 60 nm (λ ex = 280 nm; [BSA] = 1 μM; [Complexes] = 0–20 μM)
The above results indicated that red shift of the maximum emission wavelength indicates that the conformation of BSA was changed and the polarity around the tryptophan residues was increased, whereas the hydrophobicity was decreased and the microenvironment around the tyrosine residues changes during the binding process [115]. As a consequence of this, the quenching of fluorescence intensities of Trp residues was higher than that of Tyr residues, suggesting that Trp residues contributed more to the quenching of intrinsic fluorescence.
In addition to the synchronous fluorescence measurements, the same evidence was observed by three-dimensional fluorescence spectra of BSA in the presence of complexes 1 and 2. The three-dimensional fluorescence spectra measurement is an advanced analytical technique which provides detailed information about conformational change. In this work, the three-dimensional fluorescence spectra of BSA and BSA + the complex 1 (or the complex 2) solutions were observed, as shown in Fig. 14a–c. The characteristics parameters of three-dimensional fluorescence are summarized in Table 7. Peak I (λ ex = 280 nm) reveals the spectral characteristics of tryptophan and tyrosine residues. After the addition of complexes 1 and 2, the fluorescence intensity of BSA decreased from 247.6 to 174.2 for complex 1 and from 247.6 to 230.3 for complex 2. This suggests a less polar environment of both residues with almost all the hydrophobic amino-acid residues of the BSA being buried in the hydrophobic pocket. In addition, the addition of complexes 1 and 2 changed the polarity of this hydrophobic microenvironment and the conformation of the BSA [116]. Peak II (λ ex = 230 nm) reveals the fluorescence spectral behavior of the polypeptide backbone structures, which is caused by the transition of π–π* transition of the BSA [117]. After the addition of complexes 1 and 2, the fluorescence intensity of BSA decreased from 22.1 to 15.4 for complex 1 and from 22.1 to 19.6 for complex 2. These results supported the hypothesis that the interaction between BSA and complexes 1 and 2 triggered microenvironmental and conformational alterations in BSA.

Three-dimensional fluorescence spectra of BSA a BSA only b BSA + complex 1 c BSA + complex 2, Conditions: [BSA] = [Complexes] = 1 μM
Cytotoxic activity evaluation by XTT assay
The optimistic results obtained from DNA intreractions and BSA-binding studies of the water soluble, and complexes 1 and 2 encouraged us to test its cytotoxicity against selected human tumor cell lines (Caco-2, A549 and MCF-7) and healthy cells (BEAS-2B) by XTT assay. For comparison, the cytotoxicity of the known anticancer drug cisplatin has also assessed against all the above cell lines. The results were analyzed by means of cell inhibition expressed as IC50 values, and the values of complexes 1 and 2 were listed in Table 8 and in Fig. 15. These complexes exhibited higher cytotoxic effects on these cancer cells with lower IC50 values indicating their efficiency in killing the cancer cells even at low concentrations in compared with cisplatin. However, among the tested cell lines, the cytotoxic activity of complexes 1 and 2 against Caco-2 was higher than that of MCF-7 and A549. In the investigated range of concentration, cytotoxic potential was observed for complexes 1 and 2 on the Caco-2 cell line, where the IC50 values were 1.07 and 0.66 μM, respectively (Table 9). In addition, sensitivity rank of tumor cell lines of complexes 1 and 2 was Caco-2 > MCF-7 > A549. All cytotoxic results demonstrate that these results of complexes 1 and 2 are not correlated with their DNA interactions and BSA-binding results.

IC50 values of complexes 1 and 2 and cisplatin against different human tumors and healthy cells
Antimicrobial activity evaluation
In general, drug combinations have proven to be an essential feature of antimicrobial treatment due to a number of important considerations: (1) they increase activity through the use of compounds with synergistic or additive activity; (2) they thwart drug resistance; (3) they decrease required doses, reducing both cost and the chances of toxic side effects; and (4) they increase the spectrum of activity [118]. Huge success was achieved in the area of anticancer drugs and antimicrobial drugs. Some of the metal-based drugs already in market are cisplatin (anticancer drug), cardolite (myocardial imaging agent drug), silverderma (skin burn drugs marketed in Spain by Aldo Union), flammazine (skin diseases drug marketed by Durpha), and matrix metalloproteinase inhibitors (cancer and inflammatory disease marketed by British Biotech) [119]. This part of the study, \({\text{Cu}}^{ 2+ }_{{({\text{aq}})}}\), nphen, dmphen, and complexes 1 and 2 were tested against all bacterial strains and resulted in different activities. For comparison, the antimicrobial activity of the Bacitracin was also assessed against all bacterial strains. The results were analyzed by means of the diameter of inhibition zones expressed as bacteria strains and the values of complexes 1 and 2 were listed in Table 9 and Fig. 16a and b. Both new complexes showed zones of inhibition for all the seven bacterial strains, with values ranging from 12.5 to 200 μg mL−1 concentrations. \({\text{Cu}}^{ 2+ }_{{({\text{aq}})}}\) did not show any inhibition zone against all bacteria in the all concentrations. In addition, complexes 1 and 2 did not show activity against the Escherichia coli (ATCC 35218) and Klebsiellapneumoniae (ATCC 700603). On the other hand, the corresponding ligands had the weaker antibacterial activities than complexes 1 and 2. These complexes 1 and 2 indicated good zones, but complex 1 was more active than complex 2. Gram-positive bacteria Staphylococcus aureus (ATCC 25923) was also showed activity in the low concentration compared with Gram-negative bacteria. These complexes 1 and 2 indicated clear inhibition zones against Enterococcus faecalis (ATCC 29212), whereas did not in the present of nphen and dmphen. It has been reported that the copper(II) complexes had the stronger antibacterial and antifungal activities than the corresponding ligands had [120]. The antibacterial activity value of the tested complexes 1 and 2 was found to be good effect against some bacteria. Therefore, the clinical use of complexes 1 and 2 can be.

Antimicrobial activity of dmphen, nphen, complexes 1 and 2 against different bacterial species. a 100 μg/mL. b 200 μg/mL
Conclusions
Two novel mono- and dimeric copper(II) complexes have been synthesized and characterized by CHN analysis, FTIR, EPR spectroscopy, and cyclic voltammetry in this work. The crystal structures of complexes 1 and 2 were determined by single-crystal X-ray diffraction. DNA/BSA binding, DNA cleavage, cytotoxic properties, and antimicrobial activities have been investigated. The reactivity towards CT-DNA and BSA reveal that complexes 1 and 2 interact with CT-DNA by a moderate intercalation and complexes 1 and 2 bind to BSA and are thus responsible for the quenching of tryptophan fluorescence via a static quenching mechanism. The capability of cleavage of pUC19 DNA by complexes 1 and 2 indicates that at the same concentration, the promotion of efficient DNA cleavage by complex 1 without adding any external agents was attributed to a possible synergic effect between dimeric Cu(II) ions. In addition, complexes 1 and 2 exhibited prominent DNA cleavage in the presence of H2O2. The cytotoxicity of complexes 1 and 2 against different cancer cell lines (A549, Caco-2, and MCF-7) and healthy cell line (BEAS-2B) was investigated. Complexes 1 and 2 exhibit high cytotoxicity with low IC50 values and show selective cytotoxicity. In addition, complexes 1 and 2 have greater cytotoxicity activity against Caco-2 cell lines than do both other cell lines and cisplatin. The antibacterial activity of complexes 1 and 2 were found to have good effect against Escherichia coli (ATCC 35218), Escherichia coli (ATCC 25922), Enterococcus faecalis (ATCC 29212), Salmonella typhimurium (ATCC 14028), Staphylococcus aureus (ATCC 25923), Yersinia enterocolitica (ATCC 9610), and Klebsiellapneumoniae (ATCC 700603). The dimeric complex (1) was observed to be comparatively more effective than that of mononuclear complex (2), which may be due to the presence of additional copper atoms in the complexes. These initial studies are promising and may shed some light on designing new potential anticancer and antimicrobial agents in the future.
References
Ronconi L, Sadler PJ (2007) Coord Chem Rev 251:1633–1648
Cohen SM, Lippard SJ (2001) Prog Nucleic Acid Res Mol Biol 67:93–130
Lippert B (1999) Cisplatin: chemistry and biochemistry of a leading anticancer drug. Wiley-VCH, New York
Abu-Surrah AS, Kettunen M (2006) Curr Med Chem 13:1337–1357
Allardyce CS, Dyson PJ (2001) Platin Met Rev 45:62–69
Ott I, Gust R (2007) Arch Pharm Chem Life Sci 340:117–126
Jakupec MA, Galanski M, Arion VB, Hartinger CG, Keppler BK (2008) Dalton Trans 2:183–194
Yang P, Guo M (1999) Coord Chem Rev 185–186:189–211
Waddell J, Elvehjem CA, Steenbock H, Hart EB, Donk EV, Riising BM (1928) J Biol Chem 77:777–795
Waddell J, Steenbock H, Elvehjem CA, Hart EB, Donk EV (1929) J Biol Chem 83:251–260
Puiu SC, Zhou Z, White CC, Neubauer LJ, Zhang Z, Lange LE, Mansfield JA, Meyerhoff ME, Reynolds MM (2009) J Biomed Mater Res B Appl Biomater 91B:203–212
Schühle DT, Peters JA, Schatz J (2011) J Coord Chem Rev 255:2727–2745
Aldakov D, Bonnassieux Y, Geffroy B, Palacin S (2009) ACS Appl Mater Interfaces 1:584–589
Easmon J, Pürstinger G, Heinisch G, Roth T, Fiebig HH, Holzer W, Jäger W, Jenny M, Hofmann J (2001) J Med Chem 44:2164–2171
Chen Z, Zhang J, Zeng P, Zhang S, Jin C (2014) Z Anorg Allg Chem 640:1506–1513
Ruiz-Azuara L (1997) 07/628,628: RE 35,458, Feb. 18, United States Patent 1992
Ruiz-Azuara L (1996) 07/628,628: 5,576,326, United States Patent 1996
Ruiz-Azuara L (1992) 07/628,843: RE 35,458, Feb. 18 (1997). United States Patent 1992
Bravo-Gomez ME, Garcia-Ramos JC, Gracia-Mora I, Ruiz-Azuara L (2009) J Inorg Biochem 103:299–309
Alemon-Medina R, Brena-Valle M, Munoz-Sanchez JL, Gracia-Mora MI, Ruiz- Azuara L (2007) Cancer Chemother Pharmacol 60:219–228
Rivero-Muller A, De Vizcaya-Ruiz A, Plant N, Ruiz L, Dobrota M (2007) Chem Biol Interact 165:189–199
Trejo-Solis C, Palencia G, Zuniga S, Rodriguez-Ropon A, Osorio-Rico L, Luvia ST, Gracia-Mora I, Marquez-Rosado L, Sanchez A, Moreno-Garcia ME, Cruz A, Bravo-Gomez ME, Ruiz-Ramirez L, Rodriguez-Enriquez S, Sotelo J (2005) Neoplasia 7:563–574
Ruiz-Azuara L, Bravo-Gómez ME (2010) Curr Med Chem 17:3606–3615
Mejia C, Ruiz-Azuara L (2008) Pathol Oncol Res 14:467–472
Chikira M, Tomizawa Y, Fukita D, Sugizaki T, Sugawara N, Yamazaki T, Sasano A, Shindo H, Palaniandavar M, Antholine WE (2002) J Inorg Biochem 89:163–173
Asemave K, Yiase SG, Adejo SO, Anhwange BA (2011) Int J Inorg Bioinorg Chem 2:11–14
Berg JM, Tymoczko JL, Stryer L (2002) Biochemistry, 5th edn. W H Freeman and Company, New York
Ottawa JH, Apps DK (1984) Biochemistry. ELBS, London
Li NC, Doody E (1952) J Am Chem Soc 74:4184–4188
Groessl M, Terenghi M, Casini A, Elviri L, Lobinski R, Dyson PJ (2010) J Anal At Spectrom 25:305–313
İnci D, Aydin R, Yılmaz D, Gençkal HM, Vatan O, Çinkılıç N, Zorlu Y (2015) Spectrochim Acta A 136(Part B):761–770
İnci D, Aydin R, Vatan O, Yılmaz D, Gençkal HM, Zorlu Y, Cavaş T (2015) Spectrochim Acta A 145:313–324
APEX2, version 2014.1-1, Bruker, 2014, Bruker AXS Inc., Madison, WI
SAINT, version 8.34A, Bruker, 2013, Bruker AXS Inc., Madison, WI
SADABS, version 2012/2, Bruker, 2012, Bruker AXS Inc., Madison, WI
SHELXTL, version 6.14, Bruker,2000, Bruker AXS Inc., Madison, WI
Spek AL (2009) Acta Cryst D65:148–155
Macrae CF, Edgington PR, McCabe P, Pidcock E, Shields GP, Taylor R, Towler M, Streek J (2006) J Appl Cryst 39:453–457
Reichmann ME, Rice SA, Thomas CA, Doty PJ (1954) J Am Chem Soc 76:3047–3053
Addision AW, Rao TN, Reedijk J, Rijn JV, Verschoor GC (1984) J Chem Soc Dalton Trans 7:1349–1356
Chen ZL, Jiang CF, Yan WH, Liang FP, Batten SR (2009) Inorg Chem 48:4674–4684
Castineiras A, Balboa S, Carballo R, Gonzalez-Perez JM, Niclos-Gutierrez J (2007) Z Anorg Allg Chem 633:717–723
Earney JJ, Finn CPB, Najafabadi BM (1971) J Phys C Solid St Phys 4:1013–1021
Hathaway BJ, Billing DE (1970) Coord Chem Rev 5:143–207
Dudley RJ, Hathaway BJ (1970) J Chem Soc A 12:2799–2803
Mauro ED, Domiciano SM (1999) J Phys Chem Solids 60:1849–1854
Yerli Y, Kazan S, Yalçın O, Aktas B (2006) Spectrochim Acta A Mol Biomol Spectrosc 64:642–645
Yerli Y, Köksal F, Karadag A (2003) Solid State Sci 5:1319–1323
Kivelson D, Neiman R (1961) J Chem Phys 35:149–155
Ruíz P, Ortiz R, Perelló L, Alzuet G, González-Álvarez M, Liu-González M, Sanz-Ruíz F (2007) J Biol Inorg Chem 101:831–840
Halder P, Zangrando E (2010) Polyhedron 29:434–440
Ross PD, Sabramanian S (1981) Biochemistry 20(11):3096–3102
Pyle AM, Rehmann JP, Meshoyrer R, Kumar CV, Turro NJ, Barton JK (1989) J Am Chem Soc 111(8):3051–3058
Ou ZB, Lu YH, Lu YM, Chen S, Xiong YH, Zhou XH, Mao ZW, Le XY (2013) J Coord Chem 66(12):2152–2165
Zhang S, Zhou J (2008) J Coord Chem 61(15):2488–2498
Lakshmıpraba J, Arunachalam S, Solomon RV, Venuvanalıngam P (2015) J Coord Chem 68(8):1374–1386
Inci D, Aydin R, Zorlu Y (2016) J Coord Chem 69(18):2677–2696
Ganeshpandian M, Loganathan R, Ramakrishnan S, Riyasdeen A, Akbarsha MA, Palaniandavar M (2013) Polyhedron 52:924–938
Ramadevi P, Singh R, Jana SS, Devkar R, Chakraborty D (2015) J Photochem Photobiol A Chem 305:1–10
Chetana PR, Rao R, Roy M, Patra AK (2009) Inorg Chim Acta 362:4692–4698
Rao R, Patra AK, Chetana PR (2008) Polyhedron 27:1343–1352
Rao R, Patra AK, Chetana PR (2007) Polyhedron 26:5331–5338
Wu HL, Yuan JK, Bai Y, Pan GL, Wang H, Kong J, Fan XY, Liu HM (2012) Dalton Trans 41:8829–8838
Wu HL, Yuan JK, Bai Y, Pan GL, Wang H, Shu XB (2012) J Photochem Photobiol B 107:65–72
Qian W, Gu F, Gao L, Feng S, Yan D, Liao D, Cheng P (2007) Dalton Trans 10:1060–1066
Psomas G (2008) J Inorg Biochem 102(9):1798–1811
Mohamed MS, Shoukry AA, Ali AG (2012) Spectrochim Acta Part A 86:562–570
Yanmei L, Yongheng C, Zhibin O, Shi C, Chuxiong Z, Xueyi L (2012) Chin J Chem 30:303–310
Fu X-B, Zhang J-J, Liu D-D, Gan Q, Gao H-W, Mao Z-W, Le X-Y (2015) J Inorg Biochem 143:77–87
Terenzi A, Tomasello L, Spinello A, Bruno G, Giordano C, Barone G (2012) J Inorg Biochem 117:103–110
Meyer-Almes FJ, Porschke D (1993) Biochemistry 32(16):4246–4253
Karlin KD, Cohen BI, Hayes JC, Farooq A (1987) Zubieta. J Inorg Chem 26:147–153
Lee M, Rhodes AL, Wyatt MD, Forrow S, Hartley JA (1993) Biochemistry 32:4237
Lakowicz JR, Weber G (1973) Biochemistry 112(21):4161–4170
Subbaraj P, Ramu A, Raman N, Dharmaraja J (2014) J Coord Chem 67:2747–2764
Cory M, McKee DD, Kagan J, Henry DW, Miller JA (1985) J Am Chem Soc 107:2528–2536
Kelly JM, Tossi AB, McConnell DJ, OhUigin C (1985) Nucleic Acids Res 13(17):6017–6034
Goswami TK, Gadadhar S, Karande AA, Chakravarty AR (2013) Polyhedron 52:1287–1298
Seng HL, Tan KW, Maah MJ, Tan WT, Hamada H, Chikira M, Ng CH (2009) Polyhedron 28:2219–2227
Katwal R, Kaur H, Kapur BK (2013) Sci Revs Chem Commun 3(1):1–15
Powell DH, Helm L, Merbach AE (1991) J Chem Phys 95:9258–9265
Schwenk CF, Rode BM (2003) Chem Phys Chem 4:931–943
Cowan JA (2001) Curr Opin Chem Biol 5:634–642
Branum ME, Tipton AK, Zhu S, Que L Jr (2001) J Am Chem Soc 123:1898–1904
Sreedhara A, Freed JD, Cowan JA (2000) J Am Chem Soc 122:8814–8824
Deal KA, Burstyn JN (1996) Inorg Chem 35:2792–2798
Hegg EL, Burstyn JN (1998) Coord Chem Rev 173:133–165
Kenely RA, Fleming RH, Laine RM, Tse D, Winterle JS (1984) Inorg Chem 23:1870–1876
Chin J, Banaszezyk M, Jubian V, Zou XJ (1989) J Am Chem Soc 111:186–190
Chin J, Zou X (1988) J Am Chem Soc 110:223–225
Gillespie P, Ramirez F, Ugi I, Marquarding D (1993) Angew Chem Int Ed 12:91
Westheimer FH (1968) Acc Chem Res 1:70–78
Sigman DS (1990) Biochemistry 29:9097–9105
Reddy PR, Raju N (2012) Polyhedron 44:1–10
Reddy PR, Shilpa A, Raju N, Raghavaiah P (2011) J Inorg Biochem 105:1603–1612
Reddy PR, Shilpa A (2012) Chem Biodivers 9:2262–2281
Sigman DS (1986) Acc Chem Res 19(6):180–186
Hawkins MJ, Soon-Shiong P, Desaia N (2008) Adv Drug Delivery Rev 60:876–885
Paramaguru G, Kathiravan A, Selvaraj S, Venuvanalingam P, Renganathan R (2010) J Hazard Mater 175:985–991
Du N, Sheng L, Liu Z, Hu X, Xu H, Chen S (2013) Chem Cent J 7:97
Zhang LN, Wu FY, Liu AH (2011) Spectrochim Acta A 79:97–103
Lakowitcz JR (1999) Principles of fluorescence spectroscopy. Plenum Press, New York, p 237
Ware WR (1962) J Phys Chem 66:455–458
Lakowicz JR (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, New York, p 277
Hua YJ, Liu Y, Wang JB, Xiao XH, Qu SS (2004) J Pharm Biomed Anal 36:915–919
Gao H, Lei LD, Liu JQ, Kong Q, Chen XG, Hu ZD (2004) J Photochem Photobiol Part A 167:213–221
Lunardi CN, Tedesco AC, Kurth TL, Brinn IM (2003) J Photochem Photobiol Sci 2:954–959
Wang TH, Zhao ZM, Wei BZ, Zhang L, Ji L (2010) J Mol Struct 970(1–3):128–133
Tian MY, Zhang XF, Xie L, Xiang JF, Tang YL, Zhao CQ (2008) J Mol Struct 892(1–3):20–24
Wang F, Huang W, Dai ZX (2008) J Mol Struct 875(1–3):509–514
Skaikh SMT, Seetharamappa J, Kandagal PB, Ashoka S (2006) J Mol Struct 786:46–52
Cheng XX, Lui Y, Zhou B, Xiao XH, Liu Y (2009) Spectrochim Acta A 72:922–928
Miller JN (1979) Proc Anal Div Chem Soc 16(3):203–208
Guo M, Lue WJ, Li MH, Wang W (2008) Eur J Med Chem 43(10):2140–2148
Zhang G, Wang Y, Zhang H, Tang S, Tao W (2007) Pestic Biochem Physiol 87(1):23–29
Guo XJ, Sun XD, Xu SK (2009) J Mol Struct 931(1–3):55–59
Xiao Q, Huang S, Liu Y, Tian FF, Zhu JC (2009) J Fluoresc 19(2):317–326
Rizzotto M (2012) A search for antibacterial agents. Bobbarala V (ed) Chapter 5, p 73
Obaleye JA, Tella AC, Bamigboye MO (2012) A search for antibacterial agents. Bobbarala (ed) Chapter 10, p 198
Liu H, Yang W, Zhou W, Xu Y, Xie J, Li M (2013) Inorg Chim Acta 405:387–394
Acknowledgements
We thank the Research Fund of Uludag University for the financial support given to the research projects (Project Numbers OUAP (F)-2015/14 and KUAP(F)-2012/76) and the Scientific and Technological Research Council of Turkey (TUBITAK) for Domestic PhD Scholarship intended for Priority Areas of the first author (Code: 2211-C). This study is a part of doctoral thesis in progress of the first author at the Graduate School of Natural and Applied Sciences of Uludag University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
İnci, D., Aydın, R., Vatan, Ö. et al. Synthesis and crystal structures of novel copper(II) complexes with glycine and substituted phenanthrolines: reactivity towards DNA/BSA and in vitro cytotoxic and antimicrobial evaluation. J Biol Inorg Chem 22, 61–85 (2017). https://doi.org/10.1007/s00775-016-1408-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-016-1408-1