
Abstract
Significant progress has been made in recent years toward understanding the processes by which an iron mineral is deposited within members of the ferritin family of 24mer iron storage proteins, enabled by high-resolution structures together with spectroscopic and kinetic studies. These suggest common characteristics that are shared between ferritins, namely, a highly symmetric arrangement of subunits that provides a protein coat around a central cavity in which the mineral is formed, channels through the coat that facilitate ingress and egress of ions, and catalytic sites, called ferroxidase centers, that drive Fe2+ oxidation. They also reveal significant variations in both structure and mechanism amongst ferritins. Here, we describe three general types of structurally distinct ferroxidase center and the mechanisms of mineralization that they are associated with. The highlighted variation leads us to conclude that there is no universal mechanism by which ferritins function, but instead there exists several distinct mechanisms of ferritin iron mineralization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The oxygenation of the atmosphere through photosynthesis resulted in several major opportunities and challenges for life on earth, some of the most important being the bioavailability and utilization of iron. Under anaerobic conditions, iron is soluble in aqueous solvents as the Fe2+ form, and when life first evolved, was readily available to living organisms. Increasing levels of O2 resulting from the evolution of photosynthetic bacteria meant that Fe3+ became the most abundant form, and because of the very low solubility of Fe3+ in aqueous solvents, the bioavailability of iron was dramatically reduced [1]. Iron, particularly under aerobic conditions, also posed problems of toxicity due to its ability to catalyze the formation of reactive oxygen species. Nature’s answer to this dual problem of availability and toxicity was the ferritin family of iron storage proteins [1, 2], which are widely distributed in nature and found in all types of cells. Ferritins have the ability to take up and reversibly store large amounts of iron in a non-toxic form. This reduces the toxicity of iron, whilst enabling the stockpiling of this precious protein cofactor when it is in plentiful supply. Most ferritins are composed of 24 subunits, arranged to form a roughly spherical protein shell with a hollow interior. Proteins that are composed of only 12 subunits, but with the ability to synthesize an iron mineral, have been discovered and characterized. These have been coined ‘mini-ferritins’ to distinguish them from 24mer ‘maxi-ferritins’ [3]. Some mini-ferritins are also DNA-binding proteins, including the DNA-binding protein from starved Escherichia coli cells (Dps). These proteins appear to be primarily involved in resistance to oxidative stress.
All ferritins share some common general structural and mechanistic features [4]. They possess an internal cavity in which iron may be stored, channels through the protein coat that connect the inner cavity with the protein’s external environment and catalytic sites that promote the oxidation of Fe2+ to Fe3+ in the first step of formation of the ferric oxyhydroxide mineral core. The mineral cores of ferritins isolated under native conditions have variable amounts of phosphate, with Fe:P ratios ranging from 1.1:1 in some BFRs to ≥10:1 in animal ferritins (see [5] and references therein). This variation most likely reflects the level of phosphate present when the mineral was formed. Phosphate content affects the crystallinity of the mineral, with high phosphate associated with poorly crystalline cores. Phosphate also influences the kinetics of mineralization. Importantly, though, it does not appear to significantly alter mechanism, e.g., [6], and most of the mechanistic studies cited in this article were carried out in the absence of phosphate.
24mer ferritins have been the subject of intensive study for several decades and, consequently, much is known about the processes by which they form a mineral iron core. It is known that the precise characteristics of individual ferritin proteins vary significantly, particularly in the electrostatic properties of the channels and in the iron-binding sites of the catalytic centers, and several distinct iron mineralization mechanisms have been proposed. Recent studies of bacterial and human ferritins led to the proposal that all 24mer ferritins mineralize iron via a common, ‘universal’ mechanism [7]. Here, we survey the available structural and mechanistic information on 24mer ferritins to evaluate whether a universal mechanism can account for the behaviors of different ferritins. Our conclusion is that despite extensive similarities between different members of the 24mer ferritin family, a universal mechanism of mineralization does not exist.
General structural overview of 24mer ferritins
Ferritins are found in all types of cells from higher eukaryotes to bacteria. They are composed of 24 subunits that assemble together in a highly symmetric arrangement to form a dodecahedron 12 nm in diameter with a hollow center 8 nm across, see Fig. 1a. Each subunit is approx. 20 kDa in mass and consists of a four α-helix bundle, with a short fifth (E) helix at the C-terminal end, Fig. 1b. In plant ferritin, there is a ~30 residue extension at the N-terminus. Channels run through the protein coat at the symmetry axes: 8 threefold channels and 6 fourfold channels. The E helices of four subunits line the fourfold channels (Fig. 1a), while the N-terminal third of three adjacent subunits form the threefold channels (Fig. 1c). The electrostatic properties of the threefold channels are generally well conserved amongst ferritins. In vertebrates and plants, they are lined by a number of charged and polar residues and contain six carboxylate residues (one aspartate and one glutamate residue from each of the symmetry-related subunits) at their narrowest point. Binding of a variety of metal ions in the threefold channels of vertebrate ferritins, as well as effects of substitutions of carboxylate residues, indicates that these are an important access route for Fe2+ into the protein cavity [9–11]. Hydrophobic residues play an important role in stabilizing the channels [12]. The properties of the fourfold channels are less well conserved; they are largely hydrophobic in vertebrate ferritins but more polar in plant and some prokaryotic ferritins [1, 13]. Prokaryotic ferritins contain an additional type of channel, called B-channels, located between the three- and fourfold channels (Fig. 1d). These contain several carboxylate residues and could play a role in Fe2+ uptake [8, 14].

Structural features of 24mer ferritins. a Schematic representation of a 24mer ferritin viewed down one of six fourfold axis channels. b Conserved four α-helical bundle structure of the ferritin subunit, with the short fifth helix at the C-terminus. The location of the two iron-binding sites of the ferroxidase center is shown in blue. c Close-up view of one of eight threefold axis channels. d Close-up view of a B-channel, which occurs only in prokaryotic ferritins. The channel is circled for clarity. All structural figures were generated using Pymol [68]
Ferritins from vertebrates contain at least two types of subunit: Heavy (H) and Light (L) chains, which share approx. 55 % overall sequence identity and nearly 80 % identity when only residues responsible for intersubunit interactions are considered [15]. A major distinction between the two subunits is that H-chains contain residues that form a dinuclear iron site that catalyzes the oxidation of Fe2+ to Fe3+. The proportion of each subunit type varies according to the tissue of origin. Tissues that typically accumulate iron contain a high proportion of L-chain (e.g., liver and spleen ferritins contain up to 90 % L-chain), while tissues that need to minimize iron toxicity have an excess of H-chain subunits (e.g., heart and brain ferritins are approximately two-thirds H-chain) [16]. Plant ferritins are also heteropolymers of two subunit types, but these are both of the H-chain type [17].
There are two distinct types of prokaryotic ferritin: bacterioferritin (BFR) and bacterial ferritins (Ftn). The latter have now also been found in archaea and so are better described as prokaryotic non-heme ferritins to distinguish them from the heme-containing BFRs [8]. All prokaryotic ferritins appear to be homopolymers consisting of H-chain-type subunits, in which some of the key residues that constitute the eukaryotic H-chain catalytic ferroxidase center are conserved. However, they are not particularly similar to eukaryotic H-chain ferritins in terms of overall sequence similarity (~20 % identity), nor to each other as Ftn proteins share only ~10–15 % identity with BFRs [18]. Ftn and BFR proteins are often present in the same cell where, despite both being capable of storing iron, they are functionally distinct. In general, Ftn proteins appear to have a dominant role in general housekeeping iron storage, for example in E. coli [19], while BFRs appear to be more important for stress response. In some cases, Ftn and BFR proteins have dual functions in iron storage and stress response [8].
Structural variety amongst the catalytic ferroxidase centers of ferritins
Under aerobic conditions, all ferritins bind and rapidly oxidize Fe2+, depositing it as a hydrous ferric oxyhydroxide mineral core with variable phosphate content inside the protein cavity. All ferritins contain catalytic sites, called ferroxidase centers, that promote Fe2+ oxidation. The number of these sites varies depending on the subunit composition. Since prokaryotic and plant ferritins are composed of only H-chain-type subunits, they contain 24 catalytic sites (one in each subunit), while in vertebrate ferritins, only the H-chain-type subunit(s) contains a ferroxidase center. In all 24mer ferritins, the ferroxidase center comprises a dinuclear iron site as its core defining structural feature [20–22]. However, there is significant variation in the structure of the center in different ferritins. In general, as discussed below, known ferroxidase centers of 24mer ferritins can be classified as belonging to one of three distinct types: H-chain-type, Ftn-type or BFR-type.
H-chain-type ferroxidase centers
Vertebrate ferritins are the most intensively studied of all ferritins and, as described above, comprise H-chain (or H-chain-type) and L-chain subunits, though only the H-chain contains the catalytic ferroxidase center. While, H- and L-chains are also found in other eukaryotes (e.g., in insects [23]), plant ferritins are exclusively composed of H-chain-type subunits [13]. The capacity to catalyze the rapid oxidation of Fe2+ ions is dependent on the ferroxidase center. The structures of the human H-chain and frog M-chain (an H-chain-type subunit) ferroxidase centers are shown in Fig. 2a, b. The human H-chain center shown is derived from a structure containing Zn2+ at the center rather than Fe3+ [24], while the frog M-chain center actually contains Fe3+, and is the first ferritin from a higher eukaryote to be observed in an iron-bound state [20]. Each structure contains two iron sites, Fe-A and Fe-B. Fe-A is ligated by a histidine (His65 in human/His61 in frog), a monodentate glutamate (Glu27/Glu23) and a bridging glutamate (Glu62/Glu58). Fe-B in the human H-chain center is ligated by the bridging glutamate (Glu62) and a terminal glutamate (Glu107). The center is often modeled with an additional glutamate (Glu61) as a ligand to Fe-B. This is because in the structure of ferritin containing Tb3+ at the ferroxidase center [21], Glu61 was found to adopt two conformations, one bound to the metal at the B site, the other projecting toward the cavity, leading to the suggestion that this residue could be involved in iron transfer into the cavity. However, a number of other structures, including the one depicted in Fig. 1a, do not feature this residue as a ligand. Perhaps more importantly, the Fe3+-containing frog center also features only a single terminal (bidentate) glutamate (Glu103) in addition to bridging Glu58 (Fig. 2b). Although the resolution of the structure was not sufficiently high to identify a bridging oxo/hydroxo species, the short iron–iron distance of 3.1 Å is consistent with a bridged diferric center [20]. Apart from these first coordination sphere ligands, Tyr34/Tyr30 and Gln141/137 are strictly conserved amongst eukaryotic ferritins. The role of the tyrosine, which is conserved in all 24mer ferritins, is unclear. Tyrosine radical formation has been observed, but this does not appear to be an essential functional feature [25]. Another possible role for the tyrosine, as well as the glutamine, is in making hydrogen bond interactions with iron-coordinating waters.

The H-chain-type ferroxidase center. a Structure of human H-chain ferritin with Zn2+ ions bound at the center (from PDB 2CEI [24]). b Structure of frog M-chain ferritin with Fe3+ ions bound at the center (from PDB 3RBC [20]). Fe-A and Fe-B (here and in subsequent figures) refer to the two distinct Fe2+-binding sites of the dinuclear center
Ftn-type ferroxidase centers
The ferroxidase centers of Ftn proteins closely resemble those of H-chain ferritin subunits. The structure of an iron-bound form of the center in FtnA from E. coli is shown in Fig. 3a, b [26, 27]. Amino acid residue side chain coordination at site Fe-A is the same as in vertebrate ferritins, involving terminal Glu17 and His53, and bridging Glu50. Its Fe-B site is distinct, however, in that in addition to the bridging Glu50 and terminal Glu94 it has another glutamate ligand, Glu130 (this can be clearly seen in Fig. 3a). In addition, a water molecule is coordinated to Fe-A and a likely oxo- or hydroxo-species bridges the two irons, and consistent with this, the inter-iron distance was found to be 3.24 Å [27]. Importantly, the additional Glu130 ligand (compared to H-chain-type centers), is actually a bridging ligand to a third iron-binding site, Fe-C (or site C), which is also coordinated by three further glutamates, Glu49, Glu126 and Glu129 and two water molecules (Fig. 3b). The Fe-B to Fe-C inter-iron distance is ~5.8 Å, and it is noteworthy that the occupancy of the Fe-B site is low compared to that of Fe-A and Fe-C.

The Ftn-type ferroxidase center. a Structure of E. coli FtnA with Fe3+ ions bound at the center (from PDB file kindly provided by P. Artymiuk (University of Sheffield). The center is shown without Fe-C to illustrate the similarity to and difference from the H-chain center. b The same structure rotated around and with Fe-C and ligands shown. c, d Equivalent views of the eukaryotic Ftn-type ferroxidase center in P. multiseries Ftn (from PDB 4IWK [31])
Sequence comparisons revealed that Fe-C ligands are highly conserved in other prokaryotic Ftn proteins [28], and the ferroxidase centers of other structurally characterized Ftn proteins are very similar to that of the E. coli protein. These include two archaeal Ftn proteins, from Archaeoglobus fulgidis and Pyrococcus furiosus, which have been structurally characterized in an iron-loaded form [28, 29]. The Fe-A to Fe-B inter-iron distance for the A. fulgidis Ftn center was 3.18 Å, and there was clear electron density between the irons, consistent with some kind of oxo-bridge and occupancy of the site by Fe3+. The Fe-C site was also occupied, the Fe3+ at this site being 6.0 Å away from Fe-B [29]. The iron-bound P. furiosus Ftn ferroxidase center [28] featured a lower occupancy at site Fe-B compared to Fe-A, with an inter-iron distance of ~3.0 Å, shorter than in other Ftn structures but again consistent with a di-Fe3+ center. The Fe-B to Fe-C distance was 6.3 Å, slightly longer than in other Ftn proteins characterized thus far [28]. As for E. coli FtnA, site Fe-C was apparently occupied at the expense of site Fe-B.
Apart from frog ferritin, the only other eukaryotic ferritin that has been characterized in an iron-bound form is that from the pennate diatom Pseudo-nitzschia multiseries. This ferritin, along with those identified in other diatom species, appears to be composed of a single subunit type with a sequence that is distinct from other eukaryotic ferritins but, remarkably, is weakly related to prokaryotic Ftn proteins [30]. Its ferroxidase center, Fig. 3c, d, contains a Fe-A site that has the same coordinating ligands (terminal Glu15 and His51, and bridging Glu48) as those of other eukaryotic ferritins. Its Fe-B site is distinct, however, in that in addition to the bridging Glu48 and terminal Glu94 it has another glutamate ligand, Glu130. The iron–iron distance following overnight soak of crystals in Fe2+ solution was 3.6 Å, longer than that for other Ftn proteins, but still consistent with a bridged diferric center [31]. As for E. coli FtnA, Glu130 is a bridging ligand to a third iron-binding site, Fe-C (or site C). Iron at this site is additionally coordinated by Glu47 (equivalent to Glu49 in FtnA) and Glu44, and two water molecules, one of which is bridging between Fe-B and Fe-C (Fig. 3d). Thus, P. multiseries is an Ftn-type ferritin with coordination at site C similar to, but distinct from, that of prokaryotic Ftn.
The position of Glu130 appears from the structural data to be flexible between coordination at site Fe-B, Fe-C and bridging between them [31]. In general, the occupancy at Fe-C was lower than those of Fe-A and -B, and was only occupied following extended soaking times. Furthermore, two additional sites on the inner surface were observed, which may serve as nucleation site for formation of the mineral core.
BFR-type ferroxidase centers
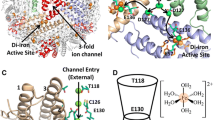
Several BFRs have been structurally characterized with iron occupying the ferroxidase center, including those from Desulfovibrio desulfuricans, E. coli, Azotobacter vinelandii and Pseudomonas aeruginosa [14, 35–37]. Unlike the other types of ferroxidase centers, BFRs have been structurally characterized in both Fe2+ and Fe3+ bound states. Although BFRs contain an intra-subunit dinuclear iron site, this is significantly different from both the H-chain and Ftn-type centers, see Fig. 4, which shows the ferroxidase center of E. coli BFR in both Fe2+-bound and Fe3+-bound forms [35]. Fe-A and Fe-B are both ligated by terminal glutamate (Glu18/Glu94 for Fe-A and Fe-B, respectively) and histidine (His54/His130) residues, and by not one but two bridging glutamate (Glu51 and Glu127) residues. This generates a highly symmetric center that is highly reminiscent of dinuclear iron centers that are found in a wide range of other (non-ferritin) proteins, including the R2 subunit of ribonucleotide reductase and the hydroxylase subunit of methane monooxygenase [38, 39]. These centers belong to class II of the dinuclear iron centers, and BFRs are the only ferritins that are members of this class. In the Fe3+-form (Fig. 4c), the inter-iron distance is 3.6 Å in the E. coli BFR structure [35], longer than in other prokaryotic ferritins, but similar to those reported for the di-Fe3+ forms of the center in D. desulfuricans (3.7 Å) and A. vinelandii (3.5 Å) BFRs [14, 36]. In each case, there is bridging electron density that is consistent with an oxygen-containing bridging species. In the Fe2+-bound state (Fig. 4a, b), there is a general increase in the Fe-A to Fe-B distance toward ~4 Å with no bridging electron density between the irons [14, 35, 36]. In some structures, His130 exhibits flexibility such that it is not always coordinated to Fe2+ bound at the Fe-B site [36].

The BFR-type ferroxidase center. a Structure of E. coli BFR with Fe2+ ions bound at the center (from PDB 3E1M [35]). b The same structure as in A rotated and zoomed out to include the inner surface site and its ligands. c Structure of E. coli BFR with Fe3+ ions bound at the center (from PDB 3E1N [35]). The oxo/hydroxo-bridge is shown in stick representation between the two irons. Tyr25 adopts one of two conformations. Note that the inner surface site is not observed in this structure due to core mineralization
There is no equivalent of Fe-C at the BFR center. However, an additional iron-binding site has been structurally characterized in E. coli BFR, in which iron (as Fe2+) is coordinated by Asp50, His46, and three water molecules [35]. This site is located on the inner surface of the subunit, facing the cavity. The distance between the inner surface site and the nearest ferroxidase center iron is 9.2 Å (Fig. 4b), and so the site is significantly more remote from Fe-A and Fe-B than is Fe-C in Ftn proteins. Despite this, Asp50 in BFR is equivalent to, but conformationally distinct from, Fe-C ligand Glu49 in FtnA.
Alignment of BFR amino acid sequences showed that while an Asp or Glu residue is almost universally found at position 50, His is only conserved at position 46 in BFRs from γ-proteobacteria and cyanobacteria; in other BFRs, it is commonly substituted, in some cases by a non-ligating residue. Given that the inner surface site has been shown to have a key functional role in E. coli BFR (see later), it must be the case that either modified and/or alternative inner surface sites exist in BFRs that do not contain the site, or that they function differently.
In addition to a distinct ferroxidase center, a further defining feature of BFRs is that they contain up to 12 hemes per 24mer, located at intersubunit binding sites [32, 33]. The heme plays a role in iron release rather than in mineralization [34], and so will not be considered further here.
Mechanistic variations in mineralization amongst ferritins
The mechanism by which ferritins deposit a ferric oxyhydroxide mineral core within their hollow centers has been studied for more than 40 years, and a vast amount of data is available on a wide range of ferritins. Some features are shared by all ferritins [1, 4, 15]. Principal amongst these are that the channels through the protein coat provide access for Fe2+ entry into the protein’s cavity, and that the catalytic ferroxidase center plays a central role in the mineralization mechanism. Additional similarities involve the actual mineral itself though how significant these are has not been established. Precipitation of Fe3+ in the cavity, which involves a phase transition from protein-associated soluble Fe3+ to the insoluble iron mineral, is likely to contribute to the rate at which Fe3+ clears the protein coat through a mass action effect. This requires further investigation but one possibility is that the phase transition ensures that Fe3+ does not get stuck in transit. Once a mineral core has formed, oxidation of Fe2+ may be catalyzed by the growing core surface, as has been established for H-chain ferritins (see below). Despite these similarities, and as discussed in the following sections, it is clear that there is significant mechanistic variation in how the mineralization reaction is driven and controlled. This is underpinned in large part by the structural diversity of the ferroxidase centers described above. However, even amongst some apparently structurally closely related centers mechanistic differences exist, such that mechanism cannot be predicted solely on the basis of the ferroxidase center type. A key consideration is the stability of Fe3+ at the ferroxidase center following oxidation of Fe2+. In some ferritins the paucity of structural data for iron-bound Fe-A and Fe-B is connected with the lability of Fe3+ from the center. In this regard, the relatively well-characterized ferroxidase center of BFRs is partly, or entirely, a consequence of the fact that Fe3+ at this center is not labile in the manner that it is in H-chain-type ferroxidase centers.
H-chain-type ferritins
Most research on eukaryotic ferritins in recent years has focused on homopolymers of H-chains. While it should be remembered that native vertebrate ferritins always contain a proportion of L-chains, these studies have been immensely useful in establishing the role of the ferroxidase center in mineralization. At low iron loadings (below or equal to that required to fill the ferroxidase centers) and under aerobic conditions, two Fe2+ ions bind at the Fe-A and Fe-B sites of the ferroxidase center, accessing it via the threefold channels [9]. Reaction with O2 results in oxidation of the two Fe2+ ions and reduction of O2 to hydrogen peroxide (a two-electron process where each Fe2+ contributes one electron, resulting in an iron-to-O2 ratio of 2:1) [40–42], see Eq. 1:
where P represents ferritin-bound iron, FC indicates iron at the ferroxidase center, and z is the net charge on the protein. This reaction proceeds via a blue diferric peroxo (DFP) intermediate [42–46], the formation and decay of which can be followed directly through its characteristic absorbance in the 650 nm region [43, 47, 48]. Although not detected in all H-chain ferritins, a DFP species has been observed in several, and so it is reasonable to assume that this is a general feature of the H-chain ferroxidase reaction, and the relative rates of its formation and decay dictate whether it can be observed. A tyrosyl radical associated with Tyr34, which lies close to the ferroxidase center, has also been detected during the ferroxidase reaction [25], but at a low level, and the importance of this is unclear because several H-chain variants exhibit wild-type level ferroxidase activity without detectable formation of such a radical.
Following oxidation, the µ-1,2-oxodiferric species resulting from Fe2+ oxidation is not stable at the ferroxidase center, but undergoes a hydrolysis reaction, leading to proton release [49] and giving [2FeOOH] which forms part of the nascent mineral, see Eq. 2:
Recent studies of H-chain ferritin, in which accessibility to complex formation with transferrin was used to indirectly monitor the location of Fe3+ in the protein, led to the suggestion that Fe3+ remains stable at the ferroxidase center following oxidation [7]. However, this is at odds with many other reports in which migration of Fe3+ out of the ferroxidase center and formation of small iron clusters have been demonstrated directly by time-resolved (including freeze-quench) Mössbauer spectroscopy for both human H-chain and frog ferritins [42, 50, 51]. The rate at which Fe3+ migrates out of the ferroxidase center into the cavity is significantly enhanced by the addition of further Fe2+, implying a displacement mechanism [7, 45]. The instability of Fe3+ at the ferroxidase center is consistent with the great difficulty in obtaining high-resolution structures of an iron-bound form of the H-chain ferroxidase center (see above).
The regeneration of the apo-form of the H-chain ferroxidase center for further rounds of Fe2+ oxidation [45, 49, 52] indicates that it is a gated iron site, facilitating transfer of iron into the cavity. Here, ‘gated’ means that transfer is coupled to ferroxidase activity, such that iron transfer can only occur following the oxidation of Fe2+ to Fe3+. Significant new insight into the pathway that Fe3+ ions take upon leaving the ferroxidase center was provided by NMR experiments in which the relaxation effects of paramagnetic Fe3+ on resonances of specific residues were detected [53]. Remarkably, the data indicated that Fe3+ takes a route along the long axis of the subunit, emerging at the fourfold axis where it enters the cavity. It was suggested that this permits cooperativity of subunits and favors the nucleation of nascent iron minerals as soon as they exit the subunit.
A further characteristic of H-chain (and also natural vertebrate ferritins with mixed H and L composition) is that once a mineral core has formed, oxidation of Fe2+ is catalyzed by the growing core surface [41, 49, 54], see Eq. 3:
where cavity indicates that the iron is located in the protein cavity but not yet part of the iron mineral. In Eq. 3, the iron-to-oxygen ratio is 4:1, suggesting that the two reaction pathways (in Eqs. 1 and 3) can be distinguished. However, H2O2 produced at the ferroxidase center can also act as an oxidant of Fe2+ at the core surface [41], and so the Fe2+:O2 ratio tends toward 4:1. Thus, once, a significant core has formed, further mineralization in H-chain-type ferritins occurs via a complex interplay of parallel oxidation pathways. The mineralization mechanism of H-chain-type ferritins is summarized in Fig. 5a.

Mechanisms of mineralization in 24mer ferritins. a Mechanism for H-chain-type ferritins describing how the ferroxidase center operates as a gated iron site (left hand side). When a mineral core is established, the surface of the mineral competes as a catalytic site. b Mechanistic schemes for Ftn-type ferritins. On the left, the Fe-C site is involved in Fe2+ oxidation, while on the right, it functions as a transit/holding site for iron oxidized at the ferroxidase center. c Mechanism for E. coli BFR-type ferritins describing how the ferroxidase center operates as a true cofactor, cycling its oxidation state as Fe2+ is oxidized in the cavity (left hand side). When a mineral core is established, the mineral itself conducts electrons toward the ferroxidase center
Ftn-type ferritins
Mineralization in Ftn proteins occurs via a mechanism related to that of H-chain ferritins but is complicated by the presence of the Fe-C site [27]. Fe2+ binding at the ferroxidase center in the presence of O2 leads to iron oxidation. A colored intermediate with absorption in the region of 600 nm was observed for E. coli FtnA, likely corresponding to a DFP species [42, 47, 55, 56]. The intermediate decays to give a μ-oxo-bridged Fe3+ dimer, which may protonate to yield a μ-hydroxo-bridged dimer [57, 58]. The Fe2+:O2 ratio for the initial oxidation reaction seems to be variable depending on the Ftn protein. In E. coli FtnA the ratio is ~3–4 [55, 56], in Ftn from the archaeon P. furiosus it is ~2–3 [59, 60] and in the Ftn from the diatom P. multiseries it is ~2 [30], indicating that the final product of O2 reduction (H2O versus H2O2) or the proportions of each generated in the different Ftn proteins are not identical. H2O2 is a more effective oxidant for Fe2+ at the ferroxidase center than O2, at least for E. coli FtnA, consistent with H2O2 produced at one center oxidizing Fe2+ ions at another, as is the case for BFR (see below). It was originally proposed that, once oxidized, iron at the Ftn ferroxidase center is unstable and, as in H-chain ferritins, migrates into the central cavity where core mineralization occurs. While this does appear to be the case, it is clear that the flux of iron through the ferroxidase center of Ftn proteins is much slower than for H-chain-type ferritins [55, 56, 59], to the extent that in some proteins, such as P. furiosus Ftn, it appears that Fe3+ is stable at the ferroxidase center until further Fe2+ is added [7, 59]. In P. multiseries Ftn, even when more Fe2+ than is needed to saturate the ferroxidase centers is added, flux through the ferroxidase center is markedly slower than observed in H-chain ferritins [31]. These properties account for why Fe3+-bound forms of several Ftn proteins have been structurally characterized (see above) and it can be concluded that they must be associated with structural differences at the ferroxidase center, which reduce the rate of Fe3+ exit relative to H-chain centers. The most obvious structural difference is the third iron site, Fe-C.
The Fe-C site appears to be of variable importance for catalytic activity in different Ftn proteins. An E. coli FtnA variant lacking site C exhibited only a small decrease in oxidation rate, but the Fe2+:O2 ratio dropped to 2 [55, 56]. Furthermore, this variant exhibited more rapid regeneration of the initial rapid oxidation phase, consistent with the proposal that the site is important for controlling iron flux [55] and without site C, the center became structurally and functionally similar to the H-chain-type center. In P. furiosus Ftn, substitution of site C ligands led to a dramatic reduction in the initial Fe2+ oxidation rate, indicating that the capacity of sites Fe-A and Fe-B to catalyze Fe2+ oxidation is dependent on Fe-C [7]. A tyrosyl radical associated with Tyr24, a conserved residue near the ferroxidase center, has been observed in both E. coli and P. furiosus Ftn proteins. This led to the suggestion that an additional, parallel mechanism could involve the oxidation of Tyr24 to a radical [7, 56]. The resulting electron would, along with three resulting from oxidation of Fe2+ ions at sites Fe-A, Fe-B and Fe-C, allow the four-electron reduction of O2 to water. The precise role of the Fe-C site in Ftn proteins remains unclear and it may be that it has distinct functions in different Ftn proteins, perhaps acting as a site of Fe2+ oxidation in some, and as a transit/holding site in others [7, 31, 56]. Mineralization mechanisms for Ftn-type ferritins are illustrated in Fig. 5b.
BFR-type ferritins
Though a number of BFRs have been structurally characterized (see above), there is less mechanistic information available for this subgroup of ferritins. The best characterized is E. coli BFR. In this protein, two Fe2+ ions bind to each ferroxidase center, most likely via a pore through the subunit that connects the center with the surrounding solution [14, 35, 37]. In the presence of O2, oxidation occurs rapidly to generate a µ-oxo/hydroxo-bridged di-Fe3+ form, with reduction of O2 to H2O2 [61, 62], see Eq. 1. H2O2 is a much better oxidant of Fe2+ at the ferroxidase center than is O2 [61, 62], and so H2O2 generated at one ferroxidase center subsequently reacts with Fe2+ at another ferroxidase center to generate a second bridged diferric center, resulting in an Fe:O2 ratio of 4:1, with overall reduction of O2 to H2O, see Eqs. 1 and 4.
It is reasonable to suggest that the oxidation reaction proceeds via a DFP species, but this has not been detected in BFR to date, probably because it decays too rapidly for detection. The resulting bridged di-Fe3+ center is stable, and a large body of data support a mechanism in which the ferroxidase center functions as a true enzyme cofactor site, driving mineral core formation by continually cycling its redox state [22, 35, 61, 63, 64], rather than transferring oxidized iron into the protein’s cavity. This distinct behavior compared to the other ferroxidase center types may result from structural differences between the centers (Figs. 2, 3, 4). This is consistent with the close similarity between the BFR ferroxidase center and the diiron sites of other well-characterized diiron enzymes, such as methane monooxygenase, that also function through redox cycling with O2 as oxidant [65].
In such a mechanism, once the ferroxidase centers are saturated, excess Fe2+ enters the central cavity, via one or more of the channels in the protein coat, and binds on the inner face of the protein coat at internal nucleation sites. One such site, involving the side chains of Asp50 and His46 [35], located approx. 10 Å from the ferroxidase center, has been structurally characterized. Here, oxidation of Fe2+ occurs, and the resulting electrons are channeled to the ferroxidase center, which is reduced back to its di-Fe2+ state. The inner surface site is within range for facile electron transfer to the ferroxidase center, but the nature of the pathway has not yet been determined. The resulting di-Fe2+ ferroxidase center can now react again with O2/H2O2, and continually cycles its oxidation state until all iron has been mineralized. As the iron mineral core builds up, Fe2+ will most likely bind to the hydrated surface of the growing mineral, with the mineral itself acting as a conducting wire to channel electrons resulting from the oxidation of the incoming Fe2+ to the ferroxidase center [66]. Substitution of either inner surface site residue did not affect the ferroxidase center oxidation reaction itself, but resulted in a ~75–80 % decrease in the rate of mineralization [35], clearly demonstrating the functional importance of the inner surface site. The fact that mineralization was not completely abolished in the absence of the inner surface site suggests that one or more alternative nucleation sites and/or electron transfer pathways exist. Studies of an engineered stable BFR subunit dimer indicated that Asp126 is also important for core mineralization but not the ferroxidase center oxidation reaction [67].
This mechanism accounts for why the BFR ferroxidase center is essential throughout mineralization and why the existing mineral core is important for determining the rate of ferroxidase center-catalyzed Fe2+ oxidation, whereby a core of ~1,200 irons gives the highest rates of further mineralization [63]. It is important to note that this mechanism does not necessarily imply that the Fe2+ ions that first bind and become oxidized at the ferroxidase center remain there throughout. Once reduced back to the di-Fe2+ form, these ions might exchange with incoming Fe2+ ions. A key feature of the mechanism, summarized in Fig. 5c, is that while the cavity is the site of Fe2+ oxidation, the ferroxidase center is the site of O2 reduction. Given that, at least in some cells, BFRs appear to function in response to oxidative stress, this separation of Fe2+ oxidation in the cavity from O2 reduction might be functionally important.
This mechanism appears not to be common to all BFRs. Studies of P. aeruginosa BfrB suggested quite different behavior, whereby the oxidized form of the ferroxidase center was not stable, and regeneration of the ferroxidase center rapid Fe2+ oxidation phase occurred [37]. Structural studies indicated the importance of the conformationally flexible His130 in gating access to, and possibly also exit from, the ferroxidase center in this protein. Thus, in some cases at least, the BFR ferroxidase center appears to function as a gated iron site (Fig. 5a).
Concluding remarks
All ferritins share certain common structural and mechanistic features. For example, they are composed of isostructural α-helical subunits that form an essentially spherical protein shell surrounding a hollow interior, and they have the capacity to take up iron into their interiors and to catalyze, via a protein-associated center, the oxidation of iron and its mineralization as ferric oxyhydroxide. The catalytic center, known as the ferroxidase center is located at an intra-subunit site within the α-helical bundle in 24mer ferritins and has as its defining feature a dinuclear iron site. The precise nature of the ferroxidase center, amongst other structural features, varies amongst different ferritins and here we have classified centers as belonging to one of three distinct types, H-chain-type, Ftn-type and BFR-type. These structural variations underpin mechanistic diversity, and it is clear that no single (universal) mechanism of iron mineralization exists that adequately describes all ferritins. The ferroxidase center plays a central role in each of the mechanisms described here. However, aside from being the site of O2 (or H2O2) reduction, the nature of that role varies. In H-chain ferritins, the center functions as a gated iron site, where the passage of iron into the protein interior is coupled to the oxidation of the iron. In BFRs, the center functions as a true enzyme cofactor, continually redox cycling as long as the Fe2+ substrate (and oxidant) is present. Ftn proteins represent another variation in which a third iron site at the ferroxidase center appears to function (in a way that differs amongst Ftn proteins) to reduce the flux of iron through the protein and therefore to slow the rate of mineralization. It is not clear why such a modified catalytic center should have evolved, though it has been suggested that it may permit iron to remain at the ligand-accessible ferroxidase center longer, which could be important for satisfying cellular iron requirements [55]. Clearly, mechanistic diversity is related to structural variation of the type described here. However, the BFR proteins from E. coli and P. aeruginosa, which appear to be structurally very similar, apparently have markedly different mechanisms of mineralization, emphasizing that the structure of the ferroxidase center, and the nature of the first coordination sphere ligands, cannot alone be used as a reliable indicator of mechanism. There must, therefore, be other factors that are important in determining the mechanism of core formation that we do not yet understand. One such factor is likely to be second coordination sphere effects which, whilst subtle for the majority of ferritin ferroxidase centers, appear to play a significant role in some cases. Despite the increasing availability of high quality structural data on ferritins from each of the classes described here, understanding the correlation of structural variation (at and remote from the catalytic center) with the observed mechanistic diversity poses a significant challenge for the future.
References
Lewin A, Moore GR, Le Brun NE (2005) Dalton Trans 3597–3610
Liu XF, Theil EC (2005) Acc Chem Res 38:167–175
Theil EC, Matzapetakis M, Liu XF (2006) J Biol Inorg Chem 11:803–810
Theil EC (2011) Curr Opin Chem Biol 15:304–311
Le Brun NE, Thomson AJ, Moore GR (1997) In metal sites in proteins and models. Springer, Berlin, pp 103–138
Aitken-Rogers H, Singleton C, Lewin A, Taylor-Gee A, Moore GR, Le Brun NE (2004) J Biol Inorg Chem 9:161–170
Ebrahimi HK, Bill E, Hagedoorn PL, Hagen WR (2012) Nat Chem Biol 8:941–948
Le Brun NE, Crow A, Murphy MEP, Mauk AG, Moore GR (2010) Biochim Biophys Acta 1800:732–744
Tosha T, Ng HL, Bhattasali O, Alber T, Theil EC (2010) J Am Chem Soc 132:14562–14569
Levi S, Santambrogio P, Corsi B, Cozzi A, Arosio P (1996) Biochem J 317:467–473
Haldar S, Bevers LE, Tosha T, Theil EC (2011) J Biol Chem 286:25620–25627
Liu XF, Jin WL, Theil EC (2003) Proc Natl Acad Sci USA 100:3653–3658
Masuda T, Goto F, Yoshihara T, Mikami B (2010) J Biol Chem 285:4049–4059
Macedo S, Romao CV, Mitchell E, Matias PM, Liu MY, Xavier AV, LeGall J, Teixeira M, Lindley P, Carrondo MA (2003) Nat Struct Biol 10:285–290
Chasteen ND, Harrison PM (1999) J Struct Biol 126:182–194
Harrison PM, Arosio P (1996) Biochim Biophys Acta 1275:161–203
Li C, Hu X, Zhao G (2009) Biochimie 91:230–239
Andrews SC (2010) Biochim Biophys Acta 1800:691–705
Abdul-Tehrani H, Hudson AJ, Chang YS, Timms AR, Hawkins C, Williams JM, Harrison PM, Guest JR, Andrews SC (1999) J Bacteriol 181:1415–1428
Bertini I, Lalli D, Mangani S, Pozzi C, Rosa C, Theil EC, Turano P (2012) J Am Chem Soc 134:6169–6176
Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JMA, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WV, Harrison PM (1991) Nature 349:541–544
Le Brun NE, Wilson MT, Andrews SC, Guest JR, Harrison PM, Thomson AJ, Moore GR (1993) FEBS Lett 333:197–202
Hamburger AE, West AP, Hamburger ZA, Hamburger P, Bjorkman PJ (2005) J Mol Biol 349:558–569
Toussaint L, Bertrand L, Hue L, Crichton RR, Declercq JP (2007) J Mol Biol 365:440–452
Chen-Barrett Y, Harrison PM, Treffry A, Quail MA, Arosio P, Santambrogio P, Chasteen ND (1995) Biochemistry 34:7847–7853
Hempstead PD, Hudson AJ, Artymiuk PJ, Andrews SC, Banfield MJ, Guest JR, Harrison PM (1994) FEBS Lett 350:258–262
Stillman TJ, Hempstead PD, Artymiuk PJ, Andrews SC, Hudson AJ, Treffry A, Guest JR, Harrison PM (2001) J Mol Biol 307:587–603
Tatur J, Hagen WR, Matias PM (2007) J Biol Inorg Chem 12:615–630
Johnson E, Cascio D, Sawaya MR, Gingery M, Schroder I (2005) Structure 13:637–648
Marchetti A, Parker MS, Moccia LP, Lin EO, Arrieta AL, Ribalet F, Murphy MEP, Maldonado MT, Armbrust EV (2009) Nature 457:467–470
Pfaffen S, Abdulqadir R, Le Brun NE, Murphy ME (2013) J Biol Chem 288:14917–14925
Andrews SC, Le Brun NE, Barynin V, Thomson AJ, Moore GR, Guest JR, Harrison PM (1995) J Biol Chem 270:23268–23274
Frolow F, Kalb AJ, Yariv J (1994) Nat Struct Biol 1:453–460
Yasmin S, Andrews SC, Moore GR, Le Brun NE (2011) J Biol Chem 286:3473–3483
Crow A, Lawson TL, Lewin A, Moore GR, Le Brun NE (2009) J Am Chem Soc 131:6808–6813
Swartz L, Kuchinskas M, Li HY, Poulos TL, Lanzilotta WN (2006) Biochemistry 45:4421–4428
Weeratunga SK, Lovell S, Yao H, Battaile KP, Fischer CJ, Gee CE, Rivera M (2010) Biochemistry 49:1160–1175
Nordlund P, Sjoberg BM, Eklund H (1990) Nature 345:593–598
Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P (1993) Nature 366:537–543
Jameson GNL, Jin W, Krebs C, Perreira AS, Tavares P, Liu XF, Theil EC, Huynh BH (2002) Biochemistry 41:13435–13443
Zhao GH, Bou-Abdallah F, Arosio P, Levi S, Janus-Chandler C, Chasteen ND (2003) Biochemistry 42:3142–3150
Bou-Abdallah F, Papaefthymiou GC, Scheswohl DM, Stanga SD, Arosio P, Chasteen ND (2002) Biochem J 364:57–63
Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH (1998) Biochemistry 37:9871–9876
Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM (1999) Biochemistry 38:5290–5295
Bou-Abdallah F, Zhao GH, Mayne HR, Arosio P, Chasteen ND (2005) J Am Chem Soc 127:3885–3893
Zhao GH, Su MH, Chasteen ND (2005) J Mol Biol 352:467–477
Zhao ZW, Treffry A, Quail MA, Guest JR, Harrison PM (1997) Dalton Trans 3977–3978
Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM (1995) Biochemistry 34:15204–15213
Yang XK, Chen-Barrett Y, Arosio P, Chasteen ND (1998) Biochemistry 37:9743–9750
Bauminger ER, Harrison PM, Hechel D, Nowik I, Treffry A (1993) Nucl Instr Meth Phys Res B 76:403–404
Pereira AS, Tavares P, Lloyd SG, Danger D, Edmondson DE, Theil EC, Huynh BH (1997) Biochemistry 36:7917–7927
Waldo GS, Theil EC (1993) Biochemistry 32:13262–13269
Turano P, Lalli D, Felli IC, Theil EC, Bertini I (2010) Proc Natl Acad Sci USA 107:545–550
Xu B, Chasteen ND (1991) J Biol Chem 266:19965–19970
Treffry A, Zhao ZW, Quail MA, Guest JR, Harrison PM (1998) FEBS Lett 432:213–218
Bou-Abdallah F, Yang H, Awomolo A, Cooper B, Woodhall MR, Andrews SC, Chasteen ND (2014) Biochemistry 53:483–495
Bauminger ER, Treffry A, Quail MA, Zhao ZW, Nowik I, Harrison PM (1999) Biochemistry 38:7791–7802
Bauminger ER, Treffry A, Quail MA, Zhao ZW, Nowik I, Harrison PM (2000) Inorg Chim Acta 297:171–180
Ebrahimi KH, Hagedoorn PL, Jongejan JA, Hagen WR (2009) J Biol Inorg Chem 14:1265–1274
Ebrahimi KH, Hagedoorn PL, Hagen WR (2013) Chembiochem 14:1123–1133
Yang X, Le Brun NE, Thomson AJ, Moore CR, Chasteen ND (2000) Biochemistry 39:4915–4923
Bou-Abdallah F, Lewin AC, Le Brun NE, Moore GR, Chasteen ND (2002) J Biol Chem 277:37064–37069
Baaghil S, Lewin A, Moore GR, Le Brun NE (2003) Biochemistry 42:14047–14056
Lawson TL, Crow A, Lewin A, Yasmin S, Moore GR, Le Brun NE (2009) Biochemistry 48:9031–9039
Le Brun NE, Andrews SC, Guest JR, Harrison PM, Moore GR, Thomson AJ (1995) Biochem J 312:385–392
Watt GD, Frankel RB, Jacobs D, Huang HQ (1992) Biochemistry 31:5672–5679
Wong SG, Tom-Yew SAL, Lewin A, Le Brun NE, Moore GR, Murphy MEP, Mauk AG (2009) J Biol Chem 284:18873–18881
DeLano WL (2002) The PyMOL molecular graphics system. DeLano Scientific, San Carlos
Acknowledgments
We thank the UK’s BBSRC for supporting our work on ferritins through Grant BB/I021884/1.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bradley, J.M., Moore, G.R. & Le Brun, N.E. Mechanisms of iron mineralization in ferritins: one size does not fit all. J Biol Inorg Chem 19, 775–785 (2014). https://doi.org/10.1007/s00775-014-1136-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-014-1136-3