
Abstract
Iron is very important in many biological processes and the ferritin protein family has evolved to store iron and to maintain cellular iron homeostasis. The deletion of the coding gene for the H subunit of ferritin leads to early embryonic death in mice and mutations in the gene for the L subunits in humans has been observed in neurodegenerative diseases, such as neuroferritinopathy. Thus, understanding how ferritin works is imperative and many studies have been conducted to delineate the molecular mechanism of ferritins and bacterioferritins. In the ferritin protein family, it is clear that a catalytic center for iron oxidation, the routes for iron to reach this center and the ability to nucleate an iron core, are common requirements for all ferritins. However, there are differences in the structural and mechanistic details of iron oxidation and mineralization. Although a common mechanism has been proposed for all ferritins, this mechanism needs to be further explored. There is a mechanistic diversity related to structural variation in the ferritin protein family. It is clear that other factors appear to affect the mechanism of iron oxidation and mineralization. This review focusses on the structural features of the ferritin protein family and its role in the mechanism of iron mineralization.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Ferritin
- Bacterial ferritin
- Bacterioferritin
- Iron biomineral
- Ferroxidase activity
- Ion channel
- Nanomaterial
- Diiron oxygenase
3.1 Introduction
Ferritin has evolved to store iron as a ferric oxyhydroxide mineral. Particulate or bound iron is encapsulated within members of the ferritin family of proteins. For living organisms the maintenance of iron homeostasis is very important, if we consider that Fe(II) is readily oxidized to Fe(III) by dioxygen and other reactive oxygen species, such as superoxide, hydrogen peroxide and water. Besides, Fe(II) reacts with hydrogen peroxide to generate the hydroxyl radical through the Fenton reaction (Boukhalfa and Crumbliss 2012), and Fe(III) is involved in the destructive catalytic cycle of Haber-Weiss (Kehrer 2000). Furthermore, the propensity of Fe(II) to react with dioxygen to generate the highly insoluble Fe(OH)3(H2O)3 (Ksp, of ∼10−38) (Boukhalfa and Crumbliss 2012) limits the bio-availability of Fe(II).
The chemical properties of iron show the importance of ferritin in cellular iron homeostasis. Besides, deletion of the coding gene for the H subunit of ferritin leads to early embryonic death in mice (Ferreira et al. 2000) and mutations in the gene of the L subunits in humans has been observed in neurodegenerative diseases such as neuroferritinopathy (Curtis et al. 2001). Furthermore, in Parkinson’s or Alzheimer’s disease (Jellinger et al. 1990) and in acquired immunodeficiency syndrome (Drakesmith et al. 2005) an increase of the ferritin expression level and the concentration of Fe(III) has been reported. Also, other roles for ferritin have been suggested (Watt 2011), including a role in immunity and autoimmunity (Recalcati et al. 2008) and in lipid metabolism (Bu et al. 2012). Thus, understanding how ferritin works is imperative, and many studies have been conducted to delineate the molecular mechanism of ferritins and bacterioferritins .
3.2 Structural Overview of Ferritins
Ferritin is a member of the four α-helix bundle structure superfamily (Andrews 2010), which also includes soluble methane monooxygenase, ribonucleotide reductase, rubrerythrin, bacterioferritin (BFR ) , DNA binding protein from starved cells (Dps), Dps-like proteins, and the recently identified archaeoferritin (Ebrahimi et al. 2012). Furthermore, ferritin is a member of the ferritin-superfamily, possessing iron-storage capacity, this family also includes bacterial ferritin and bacterioferritin (Carrondo 2003), Dps and Dps-like proteins (Haikarainen and Papageorgiou 2010; Calhoun and Kwon 2011; Wiedenheft et al. 2005) and archaeoferritin.
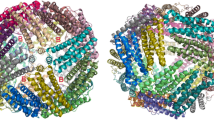
The quaternary structure of ferritin consists of 24 subunits and each subunit is characterized by four tightly packed α-helices (A–D), a loop sequence connecting the B–C helices, and a short fifth α-helix (E) that adopts an acute angle with respect to the bundle near the C-terminus tail (Le Brun et al. 2010) (see Fig. 3.1a). However, helix E adopts a different orientation in C. tepidum ferritin (CtFtn) where it forms an angle of almost 90° with respect to the four-helical bundle (Arenas-Salinas et al. 2014) (Fig. 3.1a). Despite significant differences in the primary sequences, the tertiary structures of the different ferritins are remarkably similar, however, significant differences in quaternary structure have been observed. Where Dps and Dps-like proteins form 12-mer assemblies,where ferritin, bacterial ferritin and BFR are made of 24 identical subunits (24-mer). Interestingly, BFR has a heme group between pairs of subunits (Fig. 3.1b). The primary function of the Dps and Dps-like proteins appears to be the protection of DNA against oxidative damage (Haikarainen and Papageorgiou 2010; Grant et al. 1998). Archaeoferritin is a monomeric protein that assembles upon aerobic addition of Fe(II) and its physiological function is unknown (Ebrahimi et al. 2012). This review will focus on ferritins, bacterial ferritins and bacterioferritin , as they share similar structures and properties.

Structural alignment of ferritins subunits and cartoon representation of the macromolecular assembly of ferritins. (a) Structural alignment of the subunits of EcFtnA (1EUM.pdb), HuHF (2FHA.pdb), EcBFR (3E1M.pdb) and CtFtn (4CMY.pdb) where the helix E adopts a different orientation with respect to the rest of ferritin protein family. Cartoon representation of the macromolecular assembly of HuHF (b) (2FHA.pdb), EcBFR (c) showing the HEM group in yellow (2E1M.pdb), EcFtnA (d) (1EUM.pdb) and CtFtn (e) (4CMY.pdb). The figure and the structural alignment was made with Chimera (Pettersen et al. 2004). The figure was prepared using the crystal structures available in the Protein Data Bank
3.2.1 Mechanism of Ferritin Self-Assembly
Recent studies have provided evidence that dimers are the first intermediate in the ferritin self-assembly pathway. Alanine scanning mutagenesis of E. coli bacterioferritin (EcBFR) shows that mutations at the threefold and twofold symmetry axis abolished formation of the 24-mer protein shell completely and only dimers were formed (Zhang et al. 2010). The self-assembly of human H ferritin (HuHF) was studied using a recent engineering strategy, which was termed reverse metal-templated interface redesign (Huard et al. 2013; Salgado et al. 2007, 2008). The formation of a 24-mer shell was only observed after the formation of a stable dimer. Thus, this interesting study defined a path for self-assembly of the 24-mer shell through the formation of dimers. The loop between helix B and helix C of one subunit interacts with this same loop of another subunit, generating a twofold symmetry. It has been shown that deletion of two residues in this loop in HuHF abolished formation of the 24-mer complex (Levi et al. 1989b) and a Asp80Lys mutation in this loop reduced the solubility and the stability of bull frog M ferritin (BfMF) (Bernacchioni et al. 2014). Therefore, a dimer appears to be the first intermediate during the self-assembly of the ferritin protein family and as soon as dimmers are formed, the formation of the 24-mer protein shell spontaneously occurs. Furthermore, a role of salt concentration in the mechanism of self-assembly has been described for bacterial ferritin . For example, it has been described that for Archaeoglobus fulgidus ferritin (AfFtn) self-assembly only occurs in the presence of a high salt concentration (Johnson et al. 2005; Sana et al. 2013), and it has been shown that the Apo Thermotoga maritia ferritin (TmFtn) exists largely as dimers at the cytoplasmic ionic strength of moderately halophilic bacteria (Ceci et al. 2011). In a similar way, EcBFR dissociates into dimers to the same extent as TmFtn at pH values around neutrality and 0.15 M NaCl (Andrews et al. 1995). Ceci et al. (2011) suggesting that TmFtn assembly is stabilized by iron micelles, as was also described for AfFtn after incorporation of 500 Fe/24-mer (Johnson et al. 2005), however, this characteristic has not been described in general for members of the ferritin protein family.
3.2.2 Structural Variety Amongst the Catalytic Ferroxidase Centers of Ferritins
Animal ferritins consist of two related subunits, the heavy and the light chain (H and L chain), that assemble in different proportions to form the 24-subunit protein cage (Cho et al. 2009; Toussaint et al. 2007a, b; Ha et al. 1999; Trikha et al. 1995; Granier et al. 1997). The HuHF, bacterial ferritins and BFR contain a highly conserved catalytic center called the ferroxidase center (FC), where iron is oxidized as the initial step of iron mineralization. This center possesses two iron binding sites (A and B), however, a third binding site (site C) has been found near the ferroxidase center in some Archaeal and bacterial ferritins and in BFR , as well as in mammalian ferritin (Le Brun et al. 2010; Toussaint et al. 2007a, b; Ha et al. 1999; Trikha et al. 1995; Granier et al. 1997; Masuda et al. 2010a; Hamburger et al. 2005; Stillman et al. 2001; Tatur et al. 2007; Johnson et al. 2005; Cho et al. 2009; Ceci et al. 2011; Ilari et al. 2000; Macedo et al. 2003; Ren et al. 2003; Khare et al. 2011; Pfaffen et al. 2013; Hitchings et al. 2014; Ebrahimi et al. 2012).
The 3-dimensional structure of representatives members from all three ferritin subfamilies have been determined by X-ray crystallography , from a range of species, including human (Toussaint et al. 2007a, b), frog (Ha et al. 1999; Trikha et al. 1995), horse (Granier et al. 1997), soybean (Masuda et al. 2010a), insect (Hamburger et al. 2005), E. coli (Stillman et al. 2001), Campylobacter jejuni (PDB code: 1krq; unpublished work), Pyrococcus furiosus (Tatur et al. 2007), A. fulgidus (Johnson et al. 2005), Helicobacter pylori (Cho et al. 2009), T. maritima (Ceci et al. 2011), Listeria innocua (Ilari et al. 2000), Desulfovibrio desulfuricans (Macedo et al. 2003), Bacillus brevis (Ren et al. 2003), Mycobacterium tuberculosis (Khare et al. 2011), Vibrio cholerae (PDB code:3QZ3; unpublished work), pinnate diatom (Pfaffen et al. 2013), Streptomyces coelicolor (Hitchings et al. 2014) and others. Fundamental to the available structures of the ferritin protein family, the FC has been classified as belonging to one of three distinct types: H-chain-type, Ftn-type and bacterioferritin type.
3.2.2.1 The H-Chain-Type Ferroxidase Centers
The BfMF was the first eukaryotic ferritin ferroxidase center observed in an Fe(III) bound state (Bertini et al. 2012). The Fe(III) at site A is coordinated to Glu23, Glu58, which bridges the two iron ions and His61. At site B, Fe(III) is coordinated by the bridges Glu58 and Glu103 (Bertini et al. 2012)(see Fig. 3.2a). Close to the FC, Tyr34/Tyr30 and Gln41/137 are strictly conserved amongst eukaryotic ferritins. Tyrosine radical formation has been detected in the catalytic reaction of the FC, however, the function of this tyrosine is unclear because it does not appear to be essential in the oxidation reaction (Chen-Barrett et al. 1995) (see Sect. 3.3). A similar coordination environment is observed in the FC of HuHF, derived from its crystal structure containing Zn(II) (Toussaint et al. 2007a, b). In BfMF the di-iron distance between site A and B is 3.1 Å, consistent with a bridged diferric center (Bertini et al. 2012). Recently, a third iron binding site in mammalian ferritins was identified. The analysis of the crystal structure of HuHF in presence of Tb(III), Zn(II) and Mg(II) showed the presence of three metal ion binding sites (Lawson et al. 1991; Toussaint et al. 2007a, b). Depending on the metal ion, the conformation of site C in HuHF varied and the amino acid ligands of the metal ion in site C are different: for Tb(III) Glu61 and Glu64 (Lawson et al. 1991); for Zn(II) His57 and Glu61(Toussaint et al. 2007a, b); and for Mg(II) Glu140 (Masuda et al. 2010a). Mutation of the coordinating residues of site C in HuHF decreased the rate of catalysis of Fe(II) oxidation in the FC (Masuda et al. 2010a, b). Therefore, site C also appears to be important for the catalysis of Fe(II) oxidation (see Sect. 3.3). In human L-chain ferritin (HuLF), which apparently oxidizes Fe(II) with a rate just above the rate of background oxidation of free Fe(II) by molecular oxygen (Levi et al. 1988, 1989a, b), the coordination environment of sites A and B is absent (Lawson et al. 1991; Boyd et al. 1985). This observation suggests that sites A and B are essential for fast catalysis of Fe(II) oxidation by HuHF.

Structural variety amongst the catalytic Ferroxidase centers of Ferritins. In (a) is shown the FC of BfFM (3RBC.pdb); the iron at site (a) is coordinated to Glu23 and Glu58, which bridges the two iron ions and His61. In the site (b), iron is coordinated by the bridges Glu58 and Glu103. In (b) is shown the FC of CtFtn (4CMY.pdb); the iron at site (a) is coordinated by Glu17 and His53, and bridging to Glu50. In site (b), iron is coordinated to the bridging Glu50 and terminal Glu94 and Glu130. The additional Glu130 ligand is actually a bridging ligand to a third iron-binding site (c), which is also coordinated by three further glutamates (Glu49, Glu126 and Glu129). In (c) is shown the FC of EcBFR (3E1M.pdb); iron at site (a and b) are coordinated by Glu18/Glu94 and histidine (His54/130), respectively. Additionally, iron at site (a and b) is coordinated to two bridging glutamate (Glu51 and Glu127). The figures were made with Chimera (Pettersen et al. 2004). The figure was prepared using the crystal structures available in the Protein Data Bank
In addition, three metal ion binding sites have been described in BfMF. Specifically, three Co(II) ion binding sites were observed in the X-ray structure of BfMF (Tosha et al. 2010), two sites in the middle of each subunit in the same place as the FC in HuHF and a third site with two slightly different conformations (each conformation <50% occupancy), in a position that corresponds to that of site C in HuHF. As was described to HuHF, the site directed mutation of the residues of site A, B, or C in BfMF reduced the rate of Fe(II) oxidation (Tosha et al. 2008; Behera and Theil 2014). The X-ray structure of BfMF loaded with two Fe(II) per subunit (Bertini et al. 2012) shows that the two Fe(II) are located in site A and B of the FC. In this way, the amino acids that form sites A, B and C in BfMF align with those observed in HuHF.
3.2.2.2 Bacterial Ferritin Ferroxidase Centers
The bacterial ferritin ferroxidase center closely resembles that found in H-chain ferritin. In CtFtn, the amino acid residue side chain that coordinates the iron in site A is the same as in vertebrate ferritins, involving terminal Glu17 and His53, and bridging Glu50. In site B, iron is coordinated to the bridging Glu50 and terminal Glu94 and Glu130 residues. The additional Glu130 ligand (compared to H-chain-type centers), is actually a bridging ligand to a third iron-binding site (site C), which is also coordinated by three further glutamates (Glu49, Glu126 and Glu129) (Arenas-Salinas et al. 2014) (see Fig. 3.2b). This coordination environment is similar to the one that is found in Escherichia coli (EcFtnA) (Stillman et al. 2001). Sequence comparisons have revealed that the FC ligands are highly conserved in other prokaryotic ferritins (Tatur et al. 2007). The same coordination environment that is found in EcFtnA can also be found in two Archaeal ferritins, from A. fulgidus and P. furiosus, which have been characterized in an iron loaded form (Tatur et al. 2007; Johnson et al. 2005). The iron found in P. furiosus FC (Tatur et al. 2007) shows a lower occupancy at site B compare to site A, and also shows a di-iron distance that is shorter than the distance that is seen in other ferritin structures, but which is consistent with a di-Fe(III) center. The crystal structure of AfFtn showed that the Fe-A to Fe-B distance was 3.18 Å, where an electron density was clearly observed between the irons, which would be consistent with a oxo-bridge and occupancy of Fe(III). In this case, the iron at site C is 6 Å away from Fe-B (Johnson et al. 2005), and site C is a 6.3 Å from this center. In eukaryotes, the ferritins of frog and pinnate diatom Pseudo-nitzschia multiseries have been characterized in an iron-bound form. The P. nitzschia ferritin (PmFtn) appears to be composed of a single subunit type, with a sequence that is weakly related to prokaryotic ferritins (Marchetti et al. 2009). Its FC contains a Fe-A site that has a similar coordinating environment as those of the other eukaryotic ferritins. However, its Fe-B is distinct, because it has another glutamate (Glu130) besides the bridging Glu48 and Glu94. The iron-iron distance after an overnight soak of its crystal with Fe(II) solution was 3.6 Å, longer that in other ferritins, but consistent with a bridged diferric center (Pfaffen et al. 2013). The Glu30 in EcFtnA also acts as a bridging ligand to the third iron-binding site. Therefore, PnFtn is an Ftn-type ferritin with coordination at site C, somewhat similar to what is seen in prokaryotic ferritins (Pfaffen et al. 2013).
The role of the FC in bacterial ferritin has been studied by site-directed mutagenesis, where amino acid replacement in sites A, B and C reduced the rate of Fe(II) oxidation in Pyrococcus furiosus ferritin (PfFtn) (Ebrahimi et al. 2012) and EcFtnA (Treffry et al. 1998a, b). In the crystal structure of bacterial ferritin, site A has always been found to be fully occupied with metal ions. However, in PmFtn, sites A, B or C are not occupied with metal ions (Ebrahimi et al. 2015). A full occupancy of sites A and B of the FC has been observed for BfMF and Pseudomonas aeruginosa ferritin (PaFtn) at neutral pH and for CtFtn at pH 9.0 (Arenas-Salinas et al. 2014). Taking in account the available data and the comparison of the metal ion coordination sites of the FC of different ferritins, it is possible to consider that all ferritins possess two iron binding sites in their FC (Site A and B) and a third iron binding site in the vicinity of the FC (site C). Site C of the FC in some ferritins shows a different conformation and in all structures, it is either partially occupied with metal ions or it is vacant. This suggests that this site can act as a transient iron binding site. More studies are needed to define the role of site C in the mechanism of action of ferritin. It has however been suggested that site C acts as a gateway of the FC, which is responsible for the translocation of Fe(II)/Fe(III) into or out of the FC (Ebrahimi et al. 2012)(see Sect. 3.3).
3.2.2.3 Bacterioferritin Ferroxidase Centers
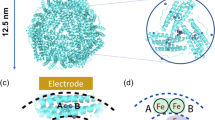
The analysis of the ferroxidase center of BFR has been possible due to several iron loaded crystal structures that have been obtained; these include those from Desulfovibrio desulfuricans, E. coli, Azotobacter vinelandii and P. aeruginosa (Macedo et al. 2003; Crow et al. 2009; Swartz et al. 2006; Weeratunga et al. 2010). BFR contains an intra-subunit di-iron site and it is different from the H-chain type and bacterial ferritin centers (see Fig. 3.2c). In EcBFR, the iron at sites A and B are coordinated by Glu18/Glu94 and His54/130, respectively. Besides, the iron at sites A and B is coordinated by two bridging glutamates (Glu51 and Glu127) (Crow et al. 2009). This symmetrical FC is similar to the dinuclear iron centers that are seen in ribonucleotide reductase and methane monooxygenase (Nordlund et al. 1990; Rosenzweig et al. 1993). Due to this characteristic, BFR belongs to class II of the dinuclear iron centers. The inter Fe(III)/Fe(III) distance is 3.6 Å in the EcBFR structure (Crow et al. 2009), longer than other prokaryotic ferritins, but similar to inter Fe(III)/Fe(III) distance of D. desulfuricans and A. vinelandii BFR (3.7 and 3.5 Å, respectively) (Macedo et al. 2003; Swartz et al. 2006). This distance increases to 4 Å in the Fe(II)/Fe(II) forms (Macedo et al. 2003; Crow et al. 2009; Swartz et al. 2006). The His130 shows flexibility and it is not always coordinated to Fe(II) at site B (Swartz et al. 2006). There is no site C in the BFR FC, which is similar to the bacterial ferritin FC center, but an additional iron binding site has been characterized in EcBFR, facing the inner cavity. In this site, the Fe(II) is coordinated by Asp50, His 46 and three water molecules (Crow et al. 2009)(Fig. 3.2c). Despite the fact that this site could be similar to site C found in bacterial ferritin, in BFR this site is located at 9.2 Å from the FC. However, the Asp50 in BFR is equivalent but not structurally identical to the site C residue Glu49 in EcFtnA. This inner iron binding site is important in the mechanism of iron mineralization in BFR (see Sect. 3.3). Another important structural feature of BFR is that it can contain up to 12 hemes per 24-mer protein, the heme group participates in iron release, as is also described in Sect. 3.3 (Andrews et al. 1995; Frolow et al. 1994).
3.2.3 Structural Variety in the Three and Fourfold Channel Amongst Ferritins (Possible Role of the B-Channel)
In the ferritin protein family, the formation of the 4-3-2 symmetrical structure of the protein shell creates channels that allow the communication of their inner cavity with the outside world. However, the analysis of the amino acid residues that form the three and fourfold channels in ferritins and bacterial ferritins show that these amino acid are not conserved among the ferritin protein family (see Tables 3.1 and 3.2). Early studies in horse spleen ferritin (HoSF) shows that Cd(II) binds to its threefold channel (Stefanini et al. 1987) and the chemical modification of Cys126 present in its threefold channel decreases the Fe(II) oxidation rate (Douglas and Ripoll 1998). Using a theoretical approach, it has been determined that the residues forming the HuHF threefold channel possess an electrostatic gradient, which allows iron to enter into its inner cavity (Douglas and Ripoll 1998; Laghaei et al. 2013). It has been shown that Zn(II) binds to the threefold channel, inhibiting the ferroxidase activity of HuHF (Bou-Abdallah et al. 2003; Toussaint et al. 2007a, b). Studies in BfMF show that Co(II) or Mg(II) are present in the threefold channel (Tosha et al. 2010). Furthermore, the flash frozen crystal of BfMF, which was aerobically soaked with Fe(II), shows the presence of this cation in the threefold channel (Bertini et al. 2012). However, similar studies show that iron was not found in the threefold or fourfold channel of PmFtn (Pfaffen et al. 2013). It has been proposed that Fe(II) reaches the FC by a diffusion controlled rate after entry into the inner cavity by the threefold channel (Bou-Abdallah et al. 2008). When residues belonging to site C are changed, a decrease Fe(II) oxidation is observed in Soybean ferritin and HuHF (Masuda et al. 2010a, b; Ensign et al. 2004). These results are in agreement with molecular dynamics (MD) simulation studies that predict that site C is present in an Fe(II) pathway to the FC on HuHF (Laghaei et al. 2013) and by the presence of Zn(II) in the crystal structure of human mitochondrial ferritin (HMFt) (Langlois et al. 2004).
Although bacterial ferritins present similar channels to those present in eukaryotic ferritin, there is no experimental evidence regarding the involvement of these channels as the iron entry route. There is only the crystal structure of HpFtn, which shows iron in its fourfold channel, which was suggest as the iron entry route to the FC and from there to the inner cavity (Cho et al. 2009). From multiple sequence alignment, it was concluded that, unlike the threefold channels in eukaryotic ferritin, the threefold channels in bacterial ferritin are hydrophobic, while the fourfold channels in some microorganism are hydrophilic. Tables 3.1 and 3.2 show that there is more variety in the amino acid composition of the threefold channel in bacterial ferritins than there is in the eukaryotic ferritins. The fourfold channel of BfMF, HuLf and HuHF, HMFt and plant ferritin are hydrophobic and all of them present a hydrophilic and negatively charged threefold channel. In contrast, prokaryotic ferritins have both hydrophilic and hydrophobic three and fourfold channel (see Tables 3.1 and 3.2). PfFtn has a hydrophilic threefold channel with a mix of positive and negative residues and a hydrophobic fourfold channel. The threefold channel of bacterioferritin B (MtBfrB) of Mycobacterium tuberculosis is similar to the PfFtn threefold channel, but MtBfrB has a hydrophobic fourfold channel. On another hand, the CtFtn has hydrophilic threefold and fourfold channels with a mix of positively and negatively charged residues. PaFtn has a hydrophilic positively charged threefold channel, but a hydrophobic fourfold channel. TmFtn and Helicobacter pylori ferritin (HpFtn) have a hydrophobic threefold channel, but a hydrophilic fourfold channel. EcFtnA, Vibrio cholerae ferritin (VcFtn) and Campylobacter jejuni ferritin (CjFtn) have hydrophobic three- and fourfold channels. An exceptional case is AfFtn, which has a hydrophobic threefold channel that forms an open pore (Johnson et al. 2005).
In general term, the quaternary structure of bacterioferritin is similar to that of ferritin and similar channels, as have been described for ferritin, are observed in bacterioferritin . However, in bacterioferritin , the threefold channels are mainly hydrophilic and the amino acids that form the fourfold channel are mainly hydrophobic. In general, three channels have been proposed as a possible pathway of Fe(II) to the ferroxidase center of bacterioferritin : the fourfold channels, B-channel and the ferroxidase channel. The ferroxidase channel was proposed based on the conformational changes of a histidine of the ferroxidase center that were observed in the crystal structures of Azotobacter vinelandii bacterioferritin (AvBFR), Pseudomonas aeruginosa bacteriferritin (PaBFR) and Desulfovibrio desulfuricans bacterioferritin (DdBFR) (Swartz et al. 2006; Weeratunga et al. 2010; Macedo et al. 2003). In this pathway, the iron enters directly to the FC, where it is oxidized and then transferred to the inner cavity. The observation of Ba(II) or Fe(III) ions at the fourfold channel in the crystal structure of AvBFR (Liu et al. 2004; Swartz et al. 2006); and K(I) in the crystal structure of PaBFR (Weeratunga et al. 2010) has been used to support the fourfold channel as the iron entry route. However, Wong et al. (2015) proposed that the B-channel is a major route for iron entry into both the ferroxidase center and the iron storage cavity of bacterioferritin . The site-directed mutagenesis of the residues belonging to the B-channel of EcBFR produced a significant decrease in the rates of initial oxidation of Fe(II) at the FC. In fact, the crystal structure of the Asp132Phe variant showed that this substitution caused a steric blockage of the B-channel (Wong et al. 2015). Studies by MD simulation predict a route where iron can reach the FC from the B-channel through site C of the FC (Weeratunga et al. 2010). However, the exact role of this site in bactrioferritin is not known. Also, Wong et al. (2015) reported that the examination of ferritins from E. coli, C. jejuni (PDB ID 1KRQ), M. tuberculosis (Nordlund et al. 1990), Vibrio cholera (PBD ID 3QZ3), T. maritime (PDB ID 1Z4A) (Salgado et al. 2008), and P. furiosus (Salgado et al. 2007) revealed that B-type channels in these structures are larger than those observed in the structure of EcBFR and should readily permit iron passage. Considering the difference in the composition of the residues belonging to the three and fourfold channels in bacterial ferritins , site C of the FC acquires an important role as the probable iron entry route to the FC from the B channels. In a similar way to what is found in eukaryotic ferritin, the mutation of site C in PfFtn decreases the Fe(II) oxidation rate (Tatur et al. 2007). In addition, the rearrangement of the side chains of two of the residues of site C (Glu51 and Glu131) observed in AfFtn is in agreement with a role for site C in the iron entry to the FC (Johnson et al. 2005). Similar conclusions were obtained when the crystal structure of BfMF in presence of Co(II), Cu(II) or Fe(II) were analyzed (Behera and Theil 2014).
3.3 Mechanistic Variations in Mineralization Amongst Ferritins
Initial studies show that in anaerobic conditions and at an iron concentration below or equal to that which is required to fill the FC of HuHF, two Fe(II) bind at sites A and B of the FC (Tosha et al. 2010). The two Fe(II) are subsequently oxidized by O2, which is reduced to hydrogen peroxide by a two-electron process where each Fe(II) contributes one electron, resulting in an iron/O2 ratio of 2:1 (Jameson et al. 2002; Zhao et al. 2003; Bou-Abdallah et al. 2002a, b) (see Fig. 3.3):

Mechanism for H-chain-type ferritins. Two Fe(II) bind at the site A and B of the FC (1). Then, the two Fe(II) are oxidized by O2 and this is reduced to hydrogen peroxide by a two-electron process where each Fe(II) contributes one electron, resulting in an iron/O2 ratio of 2:1 (2). After the initial oxidation of the Fe(II), the μ-1,2-oxodiferric species is not stable at the FC and undergoes a hydrolysis reaction, giving [2FeOOH] which forms part of the nascent inner mineral core (3)
In Eq. 3.1, P represents the iron bound to ferritin, FC represents the iron at the ferroxidase center, and z is the net charge on the protein. This reaction proceeds through a blue diferric peroxo (DFP) intermediate (Bou-Abdallah et al. 2002a, b, 2005a, b; Pereira et al. 1998; Moenne-Loccoz et al. 1999; Zhao et al. 2005). Although this has not been detected in all H-chain ferritins, a DFP species has been proposed as a general feature of the H-chain FC. During the FC reaction, a tyrosil radical close to the FC has also been detected (Chen-Barrett et al. 1995), however, the role of this radical is unclear as several H-chain variants exhibit wild-type ferroxidase activity without the detectable formation of this radical. After the initial oxidation of the Fe(II), the μ-1,2-oxodiferric species is not stable at the FC and undergoes a hydrolysis reaction, leading to proton release (Yang et al. 1998) and giving [2FeOOH] which forms part of the nascent inner mineral core (see Eq. 3.2, Fig. 3.3):
Studies with transferrin suggest that Fe(III) remains stable at the FC after its oxidation (Ebrahimi et al. 2012). However, this is in disagreement with other studies in which migration of Fe(III) out the FC and formation of small iron clusters have been demonstrated directly by time-resolved Mössbauer spectroscopy in HuHF and frog ferritin (Bou-Abdallah et al. 2002a, b; Bauminger et al. 1993; Pereira et al. 1997). The rate of Fe(III) migration from the FC into the inner cavity is enhanced by the addition of Fe(II), implying a displacement mechanism (Ebrahimi et al. 2012; Bou-Abdallah et al. 2005a, b). The latter suggests that the FC is a gated iron site, facilitating transfer of iron into the cavity (Bou-Abdallah et al. 2005a, b; Yang et al. 1998; Waldo and Theil 1993). NMR experiments indicate that Fe(III) takes a route along the long axis of the subunit, emerging at the fourfold axis, where it enters the cavity. Once a mineral core has formed, oxidation of Fe(II) is catalyzed by the growing core surface (Theil 2011; Yang et al. 1998; Xu and Chasteen 1991) (see Eq. 3.3).
Cavity indicates that the iron is located in the protein cavity but that it is not yet part of the iron mineral. In Eq. 3.3, the iron to oxygen ratio is 4:1, suggesting that the two reaction pathways (Eqs. 3.1 and 3.3) can be distinguished. Besides, the H2O2 at the FC can also act as an oxidant of Fe(II) at the core surface (Zhao et al. 2003) suggestion that the Fe(II):O2 ratio will tend toward 4:1. Therefore, once a significant core has formed, further mineralization in H-chain-type ferritin occurs via a complex of parallel oxidation pathways. However, recently Ebrahami et al. (2012) analyzed the properties of HuHF with the PfFtn, and these results were used to suggest a new mechanism of iron mineralization in ferritin. The main result of this study was the identification of a third iron binding site in HuHF, in this way both ferritins present three iron sites. In fact, these sites share similar properties, one site of high affinity and two sites of low affinity. The high affinity site was assigned to site A, and the low affinity to site B and C. The authors also proposed that the Fe(III) formed remains at the FC, because it is accessible to transferrin and through Mossbauer spectroscopy the stepwise movement of the iron was determined through the FC to the nucleation site and from there to the inner cavity. When Fe(III) reaches the site C it is less accessible and essentially unavailable in the nucleation center. Electron paramagnetic resonance experiments showed that a mixed-valent Fe(II)-Fe(III) it is produced in the FC, where addition of excess Fe(II) decreases the formation of this intermediate, suggesting that there is electron transfer from the external Fe(II) to the Fe(III) at the nucleation center. In summary, Ebrahimi et al. (2012) proposed a common mechanism for the HuHF and PfFtn. The mechanism proposed that both system possess a third Fe(II) binding site and that at least some of the oxidized Fe(III) remains at the FC upon oxidation of iron. Then, additional Fe(II) forces the Fe(III) to move from the FC to the inner cavity of ferritin. Furthermore, it was proposed as a common mechanism for all ferritins, based on their study of PfFtn and HuHF (Ebrahimi et al. 2012, 2013, 2015). In this mechanism, iron can be oxidized by two pathways (see Fig. 3.4). The first pathway is generally accepted in the literature for HuHF (Chasteen and Harrison 1999; Bou-Abdallah et al. 2002a, b, 2005a, b; Zhao et al. 2001, 2003; Yang et al. 1998) and proposes that iron is oxidized at the A and B sites of the FC and hydrogen peroxide is the product of the dioxygen reduction, giving an Fe(II)/O2 stoichiometry of 2:1, as was described above in Fig. 3.3 and Eq. 3.1. In the second pathway, iron is simultaneously oxidized at the A, B and C site of the FC, this mechanism suggests an important role for Tyr24, which form a Tyr cation radical that is neutralized by oxidation of additional Fe(II) at an unspecified site to give a Fe(II)/O2 stoichiometry of 4:1 (Fig. 3.4).

Mechanism of Fe(II) oxidation by ferritin. In this mechanism iron can distribute among of three sites, if iron bind to site A and B, the mechanism of iron oxidation follow the Fig. 3.3. If three iron bind to site A, B and C (1), two Fe(II) in the ferroxidase center are simultaneously oxidized to form a blue intermediate. The third Fe(II) either reacts with this intermediate or it is oxidized by the peroxide that is released as the blue intermediate decays. The fourth electron for complete reduction of molecular oxygen to water is proposed to be provided by the conserved tyrosine in the vicinity of the ferroxidase center (2)
The third iron site has also been proposed to function as a transit site from the FC in a number of ferritins, including EcFtnA, soybean and frog H-chain ferritin (Masuda et al. 2010a, b; Ebrahimi et al. 2012). In general, EcFtnA displays similar properties to those seen in the HuHF, both have a similar Fe(II) oxidation stoichiometry of 48 Fe(II)/shell (Zhao et al. 2001, 2003; Yang et al. 1998;Treffry et al. 1997). Iron oxidation is dependent of the functionality of sites A and B (Chasteen and Harrison 1999; Chasteen 1998; Harrison and Arosio, 1996), producing two related colored reaction intermediates (Bou-Abdallah et al. 2005a, b) and generating a tyrosil radical (Chen-Barrett et al. 1995). However, when 48 Fe(II) per EcFtnA and HuHF 24-mer are added, both proteins differ in the stoichiometry of Fe(II)/O2 oxidation, 3 and 2, respectively (Bou-Abdallah et al. 2014). Site C is highly conserved in bacterial ferritins (Stillman et al. 2003; Tatur et al. 2007; Cho et al. 2009; Ebrahimi et al. 2009; Johnson et al. 2005; Le Brun et al. 2010; Treffry et al. 1998a, b; Pereira et al. 2012). In particular, site C of EcFtnA is very important and modulates the stoichimetry and kinetics of iron oxidation. The elimination of Site C in EcFtnA produces a decrease in the Fe(II)/O2 stoichiometry from 3 to 2 for the first 48 Fe(II) added to the protein (Treffry et al. 1998b), which is similar to HuFtn. Also, the site in Eco FtnA controls the rate of regeneration of its FC, elimination of site C allows for the regeneration of their ferroxidase activity within a few hours, instead of a day that it is needed in WT E.coli FtnA (Bauminger et al. 1999).
Significantly, the iron coordination in site C in HuFH and EcFtnA are different. The C site residues Glu129 and Glu139 in EcFtnA are replaced by Lys and Ala in HuFH. However, both proteins maintain a Glu residue in site C (Glu140 and Glu106 in HuFH and EcFtnA, respectively). Another important difference is that mutations Glu140Ala and Glu140Gln in HuHF reduce the oxidation rate just to 50% (Masuda et al. 2010b; Bauminger et al. 1999), similar result can be found when the same mutations are introduced in soybean ferritin (Masuda et al. 2010a; Bauminger et al. 2000; Bou-Abdallah et al. 2014). However, the Glu129Arg, Glu129Cys and Glu129Qln mutations in PfFtn significantly reduce the oxidation rate (Ebrahimi et al. 2012; Bauminger et al. 1999; Bou-Abdallah et al. 2014). Therefore, site C can achieve different functions in these two ferritins.
In the unified mechanism described above, site C and the tyrosine radical perform essential roles (Masuda et al. 2010b; Ebrahimi et al. 2012, 2013), which are not required for rapid iron oxidation in EcFtnA (Pereira et al. 1998; Bou-Abdallah et al. 2005a, b; Treffry et al. 1998b; Bauminger et al. 1999, 2000; Stillman et al. 2003). Also, from X-ray crystallography and Mossbauer spectroscopy it has been shown that site C is not a transit site, iron does not appreciably turnover at the ferroxidase center (Bauminger et al. 1999, 2000). Bou-Abdalla and colleagues analyzed the binding of iron to the apo EcFtnA by ITC and reported that the binding of iron produced two strong, slightly endergonic binding sites and one undefined weak binding per subunit (Bou-Abdallah et al. 2014). In contrast, PfFtn shows one strong highly exergonic and two weak highly endergonic binding sites per subunit (Ebrahimi et al. 2012). Bou-Abdallah et al. (2014) proposed that, in the case of EcFtnA, once the ferroxidase center is saturated with iron, iron at site C inhibits the turnover of Fe(III) at sites A and B, they therefore serve as a redox cofactors, as was proposed for EcBFR (Baaghil et al. 2003; Crow et al. 2009) (see below).
In EcBFR, two Fe(II) ions bind to each FC, most likely via a pore through the subunit that connects the center with the surrounding solution (Macedo et al. 2003; Crow et al. 2009; Weeratunga et al. 2010). The oxidation of iron in the FC occurs rapidly in the presence of O2 to generate a μ-oxo/hydroxo bridged di-Fe(III) form, with the reduction of O2 to H2O2 (Yang et al. 2000; Bou-Abdallah et al. 2002a, b). As was described for bacterial ferritin , H2O2 is a better oxidant than O2 (Yang et al. 2000; Bou-Abdallah et al. 2002a, b). In this way, the H2O2 generated at one FC is subsequently use to oxidize Fe(II) at another FC. This means that the ratio of Fe(II):O2 is 4:1, with an overall reduction of O2 to H2O. A DPS intermediate has not been detected in BFRs to date, possibly due to rapid decay. In BFRs, the bridged di-Fe(III) center is stable, and several data support a mechanism in which the FC center functions as a true enzyme cofactor site, where it is continually cycling its redox state, see Fig. 3.5 (Le Brun et al. 1993; Crow et al. 2009; Yang et al. 2000; Baaghil et al. 2003; Lawson et al. 2009).

Mechanism of Fe(II) oxidation by bacterioferritin . Iron binds to site A and B (1) and is oxidize by with O2 or H2O2 (2). Once the FC is saturated, the excess of iron enters into the central cavity and binds on the inner face of the protein shell at internal nucleation sites, where oxidation occur (3). Then, the resulting electrons are channelled to the FC, which iron is reduced back to its Fe(II) state, and the resulting di-Fe(II) FC reacts again with O2 or H2O2 in continuous cycles until all the iron has been incorporated to the inner mineral core
This is completely different to the role of FC in ferritin and bacterial ferritin . The properties of the BFRs FC are consistent with the close similarity between these center and the di-iron sites of other well characterized di-iron enzymes, such as methane monooxygenase (Le Brun et al. 1995). The proposed mechanism for BFR , once the FC is saturated, is that the excess of iron enters into the central cavity and binds on the inner face of the protein shell at internal nucleation sites where oxidation occurs (Crow et al. 2009). Then, the resulting electrons are channeled to the FC, which is reduced back to its Fe(II) state, and the resulting di-Fe(II) FC reacts again with O2 or H2O2 in continuous cycles, until all the iron has been incorporated into the inner mineral core (Watt et al. 1992). The substitution of the surface site residue does not affect the FC activity, but does produce an 80% decrease in the rate of mineralization (Crow et al. 2009), demonstrating the importance of these surface sites in the mineralization mechanism. It has also been determined that Asp126 is important for core formation, but not for the FC reaction (Wong et al. 2009). This mechanism agrees with the observation showing that the BFR FC is essential for mineralization and that the rate of FC catalyzed Fe(II) oxidation is dependent on the mineral core (Baaghil et al. 2003). However, this mechanism cannot be common to all BFRs, as it has been determined that iron in the PaBFR FC is not stable (Weeratunga et al. 2010), suggesting that in some BFRs, FC appears to function as a gated iron site.
3.3.1 Mechanism of Iron Release in Ferritin
A mechanism for iron release in BFR has been described in which ferrodoxin can be the electron donor that reaches the mineral inner core through the heme group of EcBFR (Quail et al., 1996) (see Fig. 3.6). One the mechanisms described for iron release from ferritin includes a role for transferrin, a Fe(III) binding protein. Transferrin can scavenge iron from the FC or from the mineral core, but the rate varies between minutes and days (Ebrahimi et al. 2012). However, if this mechanism has a physiological role, it still requires further investigation. It has been suggested that this mechanism may have an important role for scavenging the Fe(III) from serum ferritin in humans (Kell and Pretorius 2014). The iron can be released from ferritin by reduction of Fe(III). In vitro, iron can be released from ferritin after chemical reduction, followed by the formation of a complex with a chelating agent for Fe(II), such as bipyridil (Funk et al. 1985; Sirivech et al. 1974) or flavorproteins (Watt et al. 1988). An interesting electrochemical study suggested a role of the PfFtn FC Fe(III) as a mediator to pass electrons from an external reducing agent to Fe(III) in the mineral core (Tatur et al. 2009). The only biologically reductants that have been found to mediate the reduction of Fe(III) under physiological conditions are the dihydroflavins (Jones et al. 1978) or flavoproteins (Watt et al. 1988). Also, it is has been proposed that mild denaturing conditions (1–10 mM Urea or 0.1 mM guanidine hydrochloride) can promote the iron release from ferritin (Liu et al. 2003). Mutations in the threefold channel amino acid residues can also increase the rate of the iron dissolution after the addition of a reducing agent (Tosha et al. 2012). However, is required further investigation to determine their physiological role.

Mechanism of Fe(III) release from bacterioferritin . Ferredoxin binds at the twofold symmetry axis of EcBFR, the Fe(II) is release after that the [2Fe-2S] cluster of ferrodoxin allow the reduction of the iron inner mineral core via an electron transfer that involves the hem group of EcBFR. The Fig. was prepared using the crystal structure of EcBFR (PDB 3E1M)
3.4 Application of the 24-mer Ferritin
Protein assemblies represent a class of highly self-organized molecules, which have the potential to be used as novel templates for nanotechnology. In this context, the ferritin proteins offer great future potential, because they form natural nanocages, where inorganic-organic hybrid materials can be synthesized (Theil and Behera 2013) (see Fig. 3.7). Ferritins present several interesting properties, as the different size of their inner cavity, the sequence variations between the ferritins protein cages from different organisms, and their different thermal and chemical stabilities. All are important when considering the use ferritin as a nanoreactor for biotechnological applications (Campan et al. 2011; Yamashita et al. 2010; Uchida et al. 2010; Maham et al. 2009). Another useful property relating to the synthesis of nano-particles is that ferritins are stable up to 80 °C, 6 M urea or guanidine hydrochloride or 1% w/v SDS (Liu and Theil 2005). Further, ferritin stability in organic solvents can be improved by derivatizing carboxylates on the external surface of the protein with long chain hydrocarbons (Liu et al. 2006; Li et al. 2007; Meldrum et al. 1992; Wade et al. 1991, 1993; Wong et al. 1999).

General methods fot the synthesis and incorporation of nanoparticles inside the ferritin inner cavity. (1) A positive charged metal diffuse into the ferritin and the nanoparticles is synthesized inside its inner cavity. (2) Intact ferritin can disassembly by incubation a pH < 2 and incubated with previously synthesized nanoparticles, then (3) the nanoparticles is stored inside ferritin inner cavity when the pH increase to 7, allowing the reassembling of the ferritin protein shell
Another useful property of ferritin is its ability to disassemble at low pH and reassemble at high pH, allowing production of different sized nanoparticles inside the protein cages (Lambet et al. 2010)(see Fig. 3.7). These properties have been used in the synthesis and delivery of magnetic resonance imaging (MRI) contrast agents (Sana et al. 2012; Li et al. 2012; Wood 2011; Terashima et al. 2011; Ziv et al. 2010) and for drug delivery and catalysis (Maham et al. 2009; Zheng et al. 2010; Xing et al. 2009; Mann 2009; Abe et al. 2009). In vitro, purified ferritin cages can incorporate a variety of metal ions (Fe, Au, Pd, Rh, Pt, Ni, Cr, Cd, Ti, Tb, Co, Cu and Zn) (Tosha et al. 2010; Uchida et al. 2010; Iwahori and Yamashita 2008). Also, it has been possible to synthesize nano-particles of gold, silver, lead, copper and nickel after incubation of apo-ferritins with ions, followed by a reducing agent (Uchida et al. 2010). Usefully, the engineering of additional metal binding sites has allowed the synthesis of silver and gold nanoparticles (Butts et al. 2008). Taking advantage of the thermal resistance of PfFtn, it has been possible to synthesize magnetite (Uchida et al. 2010). The apo-form of ferritin has an inner cavity that could be used to entrap not just metallic nanoparticle derivatives, but also organic molecules. Thus, ferritin has been reported while containing hydrophilic drugs, imaging agents (MRI and fluorophores) (Terashima et al. 2011; Kitagawa et al. 2012; Flenniken et al. 2009), radionuclides or nuclear medicines (Maham et al. 2009), anti-tumor drugs (Xing et al. 2009; Yang et al. 2007a, b) and DNA probes (Maham et al. 2009). Furthermore, genetic or chemical modification of the surface of ferritin could help to target these complexes to specific cell types, thereby improving their future use in nano-medicine . An example of the use of ferritin in nano-medicine is the development of ferritin with the peptide RGD-4 attached to its surface. This allows the specific binding of this complex to integrins v3 and v5 (Terashima et al. 2011; Kitagawa et al. 2012), which can be used for the imaging of vascular inflammation and angiogenesis and for affecting the metabolism of C32 human melanoma cells.
In an indirect method, the superparamagnetism of the inner core of ferritin can be used as an endogenous enhancer of the transverse nuclear magnetic relaxation of water. Thus it has been used to determine iron levels associated with hypertransfusion treatments of sickle cell anemia, thalassemia, and in hemochromatosis and Alzheimer disease (Wood 2011). On another hand, a similar approach, which was described earlier, has been used to check transgene expression (Ono et al. 2009). It has been reported that improvement of the MRI contrasting ability of ferritin complexes occurs upon an increase of the iron biomineral or by entrapment of synthetic magnetite or other nanoparticles (Li et al. 2012; Terashima et al. 2011; Uchida et al. 2008). Another application of ferritin as nano-reactor is in nano-catalysis and nano-electronics. Thus, ferritins have been used as sites for organometallic catalysis, to produce polymers of defined molecular weight and with a narrow size distribution (Wang et al. 2011; Takezawa et al. 2011; Abe et al. 2009). In this way, ferritin cages have been used as templates for metal oxide nanoparticles and metal oxide semiconductors and also to produce building blocks for the fabrication of electronic devices, such as the floating nanodot memory device or thin film transistor flash memory (Yamashita et al. 2010; Uchida et al. 2010).
3.5 Concluding Remarks
In the ferritin protein family, it is clear that a catalytic center for iron oxidation, the routes for iron to reach this center and the ability to nucleate an iron core are common requirements for all ferritins. However, there are differences in the structural and mechanistic details of iron oxidation and mineralization. Therefore, ferritins have been classified in three distinct types, H-chain-type, Ftn-type and BFR -type. Although a common mechanism has been proposed for all ferritins, this mechanism needs to be further explored. There is a mechanistic diversity that is related to structural variation in the ferritin protein family. For example, the highly related BFR from E. coli and P. aeruginosa have different mechanisms of mineralization, suggesting that just the first coordination sphere ligands do not allow the prediction of a mechanism. It is clear that other factors appear to affect the mechanism of iron oxidation and mineralization. A significant challenge for the future is to correlate the high quality structural data on ferritins with the biochemical diversity observed in the ferritin protein family. Further studies should contribute to our understanding of the role of this protein family in iron homeostasis and be used to develop treatments for iron metabolism disorders and new biotechnological applications.
Abbreviations
- AfFtn:
-
Archaeoglobus fulgidus ferritin
- AvBFR:
-
Azotobacter vinelandii bacterioferritin
- BFR:
-
Bacterioferritin
- BfMF:
-
Bull frog M ferritin
- CjFtn:
-
Campylobacter jejuni ferritin
- CtFtn:
-
Chlorobium tepidum ferritin
- DdBFR:
-
Desulfovibrio desulfuricans bacterioferritin
- DFP:
-
Diferric peroxo
- Dps:
-
DNA binding protein from starved cells
- EcBFR:
-
Escherichia coli bacterioferritin
- EcFtnA:
-
Escherichia coli ferritin A
- FC:
-
ferroxidase center
- HpFtn:
-
Helicobacter pylori ferritin
- HoSF:
-
Horse spleen ferritin
- HuHF:
-
Human H ferritin
- HuLF:
-
Human L ferritin
- HMFt:
-
human mitochondrial ferritin
- ITC:
-
Isothermal titration calorimetry
- MRI:
-
Magnetic resonance imaging
- MD:
-
Molecular dynamics
- MtbBFrB:
-
Mycobacterium tuberculosis bacterioferritin B
- NMR:
-
Nuclear magnetic resonance
- PaFtn:
-
Pseudomonas aeruginosa ferritin
- PaBFR:
-
Pseudomonas aeruginosa bacteriferritin
- PfFtn:
-
Pyrococcus furiosus ferritin
- PmFtn:
-
Pseudo-nitzschia ferritin
- TmFtn:
-
Thermotoga maritima ferritin
- VcFtn:
-
Vibrio cholerae ferritin
References
Abe S, Hirata K, Ueno T, Morino K, Shimizu N, Yamamoto M, Takata M, Yashima E, Watanabe Y (2009) Polymerization of phenylacetylene by rhodium complexes within a discrete space of apo-ferritin. J Am Chem Soc 131:6958–6960
Andrews SC (2010) The Ferritin-like superfamily: evolution of the biological iron storeman from a rubrerythrin-like ancestor. Biochim Biophys Acta 1800:691–705
Andrews SC, Le Brun NE, Barynin V, Thomson AJ, Moore GR, Guest JR, Harrison PM (1995) Site-directed replacement of the coaxial heme ligands of bacterioferritin generates heme-free variants. J Biol Chem 270:23268–23274
Arenas-Salinas M, Townsend PD, Brito C, Márquez V, Marabolli V, Gonzalez-Nilo F, Matias C, Watt RK, López-Castro JD, Domínguez-Vera J, Pohl E, Yévenes A (2014) The crystal structure of ferritin from Chlorobium tepidum reveals a new conformation of the 4-fold channel for this protein family. Biochimie 106:39–47
Baaghil S, Lewin A, Moore GR, Le Brun NE (2003) Core formation in Escherichia coli bacterioferritin requires a functional ferroxidase center. Biochemistry 42:14047–14056
Bauminger ER, Harrison PM, Hechel D, Nowik I, Treffry A (1993) The function of H and L chains in iron sequestration in ferritin. Nucl Inst Methods Phys Res B 76:403–404
Bauminger ER, Treffry A, Quail MA, Zhao ZW, Nowik I, Harrison PM (1999) Stages in iron storage in the ferritin of Escherichia coli (EcFtnA): analysis of Mössbauer spectra reveals a new intermediate. Biochemistry 38:7791–7802
Bauminger ER, Treffry A, Quail MA, Zhao Z, Nowik I, Harrison PM (2000) Metal binding at the active centre of the ferritin of Escherichia coli (EcFtnA). A Mössbauer spectroscopic study. Inorg Chim Acta 297:171–180
Behera RK, Theil EC (2014) Moving Fe2+ from ferritin ion channels to catalytic OH centers depends on conserved protein cage carboxylates. Proc Natl Acad Sci U S A 111:7925–7930
Bernacchioni C, Ghini V, Pozzi C, Di Pisa F, Theil EC, Turano P (2014) Loop electrostatics modulates the intersubunit interactions in ferritin. ACS Chem Biol 9(11):2517–2525. doi:10.1021/cb500431r
Bertini I, Lalli D, Mangani S, Pozzi C, Rosa C, Theil EC, Turano P (2012) Structural insights into the ferroxidase site of ferritins from higher eukaryotes. J Am Chem Soc 134:6169–6176
Bou-Abdallah F, Lewin AC, Le Brun NE, Moore GR, Chasteen ND (2002a) Iron detoxification properties of Escherichia coli bacterioferritin. Attenuation of oxyradical chemistry. J Biol Chem 277:37064–37069
Bou-Abdallah F, Papaefthymiou GC, Scheswohl DM, Stanga SD, Arosio P, Chasteen ND (2002b) mu-1,2-Peroxobridged di-iron(III) dimer formation in human H-chain ferritin. Biochem J 364:57–63
Bou-Abdallah F, Arosio P, Levi S, Janus-Chandler C, Chasteen ND (2003) Defining metal ion inhibitor interactions with recombinant human H- and L-chain ferritins and site-directed variants: an isothermal titration calorimetry study. J Biol Inorg Chem 8:489–497
Bou-Abdallah F, Zhao GH, Mayne HR, Arosio P, Chasteen ND (2005a) Origin of the unusual kinetics of iron deposition in human H-chain ferritin. J Am Chem Soc 127:3885–3893
Bou-Abdallah F, Woodhall MR, Velásquez-Campoy A, Andrews SC, Chasteen ND (2005b) Thermodynamic analysis of ferrous ion binding to Escherichia coli ferritin EcFtnA. Biochemistry 44:13837–13846
Bou-Abdallah F, Zhao G, Biasiotto G, Poli M, Arosio P, Chasteen ND (2008) Facilitated diffusion of iron(II) and dioxygen substrates into human H-chain ferritin. A fluorescence and absorbance study employing the ferroxidase center substitution Y34W. J Am Chem Soc 130:17801–17811
Bou-Abdallah F, Yang H, Awomolo A, Cooper B, Woodhall MR, Andrews SC, Chasteen ND (2014) Functionality of the three-site ferroxidase center of Escherichia coli bacterial ferritin (EcFtnA). Biochemistry 53:483–495
Boukhalfa H, Crumbliss AL (2012) Chemical aspects of siderophore mediated iron transport. BioMetals 15:325–339
Boyd D, Vecoli C, Belcher DM, Jain SK, Drysdale JW (1985) Structural and functional relationships of human ferritin H and L chains deduced from cDNA clones. J Biol Chem 260:11755–11761
Bu W, Liu R, Cheung-Lau JC, Dmochowski IJ, Loll PJ, Eckenhoff RG (2012) Ferritin couples iron and fatty acid metabolism. FASEB J 26:2394–2400
Butts CA, Swift J, Kang SG, Di Costanzo L, Christianson DW, Saven JG, Dmochowski IJ (2008) Directing noble metal ion chemistry within a designed ferritin protein. Biochemistry 47:12729–12739
Calhoun LN, Kwon YM (2011) Structure, function and regulation of the DNA-binding protein Dps and its role in acid and oxidative stress resistance in Escherichia coli: a review. J Appl Microbiol 110:375–386
Campan M, Lionetti V, Aquaro GD, Forini F, Matteucci M, Vannucci L, Chiuppesi F, Di Cristofano C, Faggioni M, Maioli M, Barile L, Messina E, Lombardi M, Pucci A, Pistello M, Recchia FA (2011) Ferritin as a reporter gene for in vivo tracking of stem cells by 1.5-T cardiac MRI in a rat model of myocardial infarction. Am J Physiol Heart Circ Physiol 300:H2238–H2250
Carrondo MA (2003) Ferritins, iron uptake and storage from the bacterioferritin viewpoint. EMBO J 22:1959–1968
Ceci P, Forte E, Di Cecca F, Fornara M, Chienacone E (2011) The characterization of Thermotoga marítima ferritin reveals an unusual subunit dissociation behavior and efficient DNA protection form iron-mediated oxidative stress. Extremophiles 15:431–439
Chasteen ND (1998) Ferritin. Uptake, storage, and release of iron. In: Sigel H, Sigel A (eds) Metal ions in biological systems, vol 35. Marcel Dekker Inc., New York, pp 479–514
Chasteen ND, Harrison PM (1999) Mineralization in ferritin: an efficient means of iron storage. J Struct Biol 126:182–194
Chen-Barrett Y, Harrison PM, Treffry A, Quail MA, Arosio P, Santambrogio P, Chasteen ND (1995) Tyrosyl radical formation during the oxidative deposition of iron in human apoferritin. Biochemistry 34:7847–7853
Cho KJ, Shin HJ, Lee JH, Kim KJ, Park SS, Lee Y, Lee C, Park SS, Kim KH (2009) The crystal structure of ferritin from Helicobacter pylori reveals unusual conformational changes for iron uptake. J Mol Biol 390:83–98
Crow A, Lawson TL, Lewin A, Moore GR, Le Brun NE (2009) Structural basis for iron mineralization by bacterioferritin. J Am Chem Soc 131:6808–6813
Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP (2001) Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet 28:350–354
Douglas T, Ripoll DR (1998) Calculated electrostatic gradients in recombinant human H-chain ferritin. Protein Sci 7:1083–1091
Drakesmith H, Chen N, Ledermann H, Screaton G, Townsend A, Xu XN (2005) HIV-1 Nef down-regulates the hemochromatosis protein HFE, manipulating cellular iron homeostasis. Proc Natl Acad Sci U S A 102:11017–11022
Ebrahimi KH, Hagedoorn PL, Jongejan JA, Hagen WR (2009) Catalysis of iron core formation in Pyrococcus furiosus ferritin. J Biol Inorg Chem 14:1265–1274
Ebrahimi KH, Bill E, Hagedoorn PL, Hagen WR (2012) The catalytic center of ferritin regulates iron storage via Fe(II)eFe(III) displacement. Nat Chem Biol 8:941–948
Ebrahimi KH, Hagedoorn PL, Hagen WR (2013) A conserved tyrosine in ferritin is a molecular capacitor. Chem Bio Chem 14:1123–1133
Ebrahimi KH, Hagedoom PL, Hagne W (2015) Unity in the biochemistry of the iron-storage proteins ferritin and bacterioferritin. Chem Rev 115:295–326
Ensign D, Young M, Douglas T (2004) Photocatalytic synthesis of copper colloids from CuII by the ferrihydrite core of ferritin. Inorg Chem 43:3441–3446
Ferreira C, Bucchini D, Martin ME, Levi S, Arosio P, Grandchamp B, Beaumont C (2000) Early embryonic lethality of H ferritin gene deletion in mice. J Biol Chem 275:3021
Flenniken ML, Uchida M, Liepold LO, Kang S, Young MJ, Douglas T (2009) A library of protein cage architectures as nanomaterials. Curr Top Microbiol Immunol 327:71–93
Frolow F, Kalb AJ, Yariv J (1994) Structure of a unique twofold symmetric haem-binding site. Nat Struct Biol 1:453–460
Funk F, Lenders JP, Crichton RR, Schneider W (1985) Reductive mobilisation of ferritin iron. Eur J Biochem 152:167–172
Granier T, Gallois B, Dautant A, Langlois d’Estaintot B, Precigoux G (1997) Comparison of the structures of the cubic and tetragonal forms of horse-spleen apoferritin. Acta Crystallogr D Biol Crystallogr 53:580-587
Grant R, Filman D, Finkel S, Kolter R, Hogle J (1998) The crystal structure of Dps, a ferritin homolog that binds and protects DNA. J Nat Struct Mol Biol 5:294–303
Ha Y, Shi D, Small GW, Theil EC, Allewell NM (1999) Crystal structure of bullfrog M ferritin at 2.8 A resolution: analysis of subunit interactions and the binuclear metal center. J Biol Inorg Chem 4:243–256
Haikarainen T, Papageorgiou A (2010) Dps-like proteins: structural and functional insights into a versatile protein family. Cell Mol Life Sci 67:341–351
Hamburger AE, West AP Jr, Hamburger ZA, Hamburger P, Bjorkman PJ (2005) Crystal structure of a secreted insect ferritin reveals a symmetrical arrangement of heavy and light chains. J Mol Biol 349:558–569
Harrison PM, Arosio P (1996) The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1275:161–203
Hitchings MD, Townsend P, Pohl E, Facey PD, Jones DH, Dyson PJ, Del Sol R (2014) Tale of tails: deciphering the contribution of terminal tails to the biochemical properties of two Dps proteins from Streptomyces coelicolor. Cell Mol Life Sci 24:4911–4926
Huard DJ, Kane KM, Tezcan FA (2013) Re-engineering protein interfaces yields copper-inducible ferritin cage assembly. Nat Chem Biol 9:169–176
Ilari A, Stefanini S, Chiancone E, Tsernoglou D (2000) The dodecameric ferritin from Listeria innocua contains a novel intersubunit iron-binding site. Nat Struct Biol 7:38–43
Iwahori K, Yamashita I (2008) Size-controlled one-pot synthesis of fluorescent cadmium sulfide semiconductor nanoparticles in an apoferritin cavity. Nanotechnology 19:495601
Jameson GNL, Jin W, Krebs C, Perreira AS, Tavares P, Liu XF, Theil EC, Huynh BH (2002) Stoichiometric production of hydrogen peroxide and parallel formation of ferric multimers through decay of the diferric-peroxo complex, the first detectable intermediate in ferritin mineralization. Biochemistry 41:13435–13443
Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MJ (1990) Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect 2:327–340
Johnson E, Cascio D, Sawaya MR, Gingery M, Schröder I (2005) Crystal structures of a tetrahedral open pore ferritin from the hyperthermophilic archaeon Archaeoglobus fulgidus. Structure 13:637–648
Jones T, Spencer R, Walsh C (1978) Mechanism and kinetics of iron release from ferritin by dihydroflavins and dihydroflavin analogues. Biochemistry 17:4011–4017
Kehrer P (2000) The Haber–Weiss reaction and mechanisms of toxicity. Toxicology 149:43–50
Kell DB, Pretorius E (2014) Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 6:748–773
Khare G, Gupta V, Nangpal P, Gupta RK, Sauter NK, Tyagi AK (2011) Ferritin structure from Mycobacterium tuberculosis: comparative study with homologues identifies extended C-terminus involved in ferroxidase activity. PLoS One 6:e18570
Kitagawa T, Kosuge H, Uchida M, Dua MM, Iida Y, Dalman RL, Douglas T, McConnell MV (2012) RGD-conjugated human ferritin nanoparticles for imaging vascular inflammation and angiogenesis in experimental carotid and aortic disease. Mol Imaging Biol 14:315–324
Laghaei R, Evans DG, Coalson RD (2013) Metal binding sites of human H-chain ferritin and iron transport mechanism to the ferroxidase sites: a molecular dynamics simulation study. Proteins 81:1042–1050
Langlois d’Estaintot B, Santambrogio P, Granier T, Gallois B, Chevalier JM, Précigoux G, Levi S, Arosio P (2004) Crystal structure and biochemical properties of the human mitochondrial ferritin and its mutant Ser144Ala. J Mol Biol 340:277–293
Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JMA, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WV, Harrison PM (1991) Solving the structure of human H ferritin by genetically engineering intermolecular crystal contacts. Nature 349:541–544
Lawson TL, Crow A, Lewin A, Yasmin S, Moore GR, Le Brun NE (2009) Monitoring the iron status of the ferroxidase center of Escherichia coli bacterioferritin using fluorescence spectroscopy. Biochemistry 48:9031–9039
Le Brun NE, Wilson MT, Andrews SC, Guest JR, Harrison PM, Thomson AJ, Moore GR (1993) Kinetic and structural characterization of an intermediate in the biomineralization of bacterioferritin. FEBS Lett 333:197–202
Le Brun NE, Andrews SC, Guest JR, Harrison PM, Moore GR, Thomson AJ (1995) Identification of the ferroxidase centre of Escherichia coli bacterioferritin. Biochem J 312:385–392
Le Brun NE, Crow A, Murphy ME, Mauk AG, Moore GR (2010) Iron core mineralization in prokaryotic ferritins. Biochim Biophys Acta 1800:732–744
Levi S, Luzzago A, Cesareni G, Cozzi A, Franceschinelli F, Albertini A, Arosio PJ (1988) Mechanism of ferritin iron uptake: activity of the H-chain and deletion mapping of the ferro-oxidase site. A study of iron uptake and ferro-oxidase activity of human liver, recombinant H-chain ferritins, and of two H-chain deletion mutants. Biol Chem 263:18086–18092
Levi S, Salfeld J, Franceschinelli F, Cozzi A, Dorner MH, Arosio P (1989a) Expression and structural and functional properties of human ferritin L-chain from Escherichia coli. Biochemistry 28:5179–5184
Levi S, Luzzago A, Franceschinelli F, Santambrogio P, Cesareni G, Arosio P (1989b) Mutational analysis of the channel and loop sequences of human ferritin H-chain. Biochem J 264:381–388
Li M, Viravaidya C, Mann S (2007) Polymer-mediated synthesis of ferritin-encapsulated inorganic nanoparticles. Small 3:1477–1481
Li K, Zhang ZP, Luo M, Yu X, Han Y, Wei HP, Cui ZQ, Zhang XE (2012) Multifunctional ferritin cage nanostructures for fluorescence and MR imaging of tumor cells. Nanoscale 4:188–193
Liu X, Theil EC (2005) Ferritins: dynamic management of biological iron and oxygen chemistry. Acc Chem Res 38:167–175
Liu X, Jin W, Theil EC (2003) Opening protein pores with chaotropes enhances Fe reduction and chelation of Fe from the ferritin biomineral. Proc Natl Acad Sci U S A 100:3653–3658
Liu HL, Zhou HN, Xing WM, Zhao JF, Li SX, Huang JF, Bi RC (2004) 2.6 A resolution crystal structure of the bacterioferritin from Azotobacter vinelandii. FEBS Lett 573:93–98
Liu G, Wang J, Wu H, Lin Y (2006) Versatile apoferritin nanoparticle labels for assay of protein. Anal Chem 78:7417–7423
Macedo S, Romao CV, Mitchell E, Matias PM, Liu MY, Xavier AV, LeGall J, Teixeir M, Lindley P, Carrondo MA (2003) The nature of the di-iron site in the bacterioferritin from Desulfovibrio desulfuricans. Nat Struct Biol 10:285–290
Maham A, Tang Z, Wu H, Wang J, Lin Y (2009) Protein-based nanomedicine platforms for drug delivery. Small 5:1706–1721
Mann S (2009) Self-assembly and transformation of hybrid nano-objects and nanostructures under equilibrium and non-equilibrium conditions. Nat Mater 8:781–792
Marchetti A, Parker MS, Moccia LP, Lin EO, Arrieta AL, Ribalet F, Murphy MEP, Maldonado MT, Armbrust EV (2009) Ferritin is used for iron storage in bloom-forming marine pennate diatoms. Nature 457:467–470
Masuda T, Goto F, Yoshihara T, Mikami B (2010a) Crystal structure of plant ferritin reveals a novel metal binding site that functions as a transit site for metal transfer in ferritin. J Biol Chem 285:4049–4059
Masuda T, Goto F, Yoshihara T, Mikami B (2010b) The universal mechanism for iron translocation to the ferroxidase site in ferritin, which is mediated by the well conserved transit site. Biochem Biophys Res Commun 400:94–99
Meldrum FC, Heywood BR, Mann S (1992) Magnetoferritin: in vitro synthesis of a novel magnetic protein. Science 257(5069):522–523
Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM (1999) The ferroxidase reaction of ferritin reveals a diferric mu-1,2 bridging peroxide intermediate in common with other O2-activating non-heme diiron proteins. Biochemistry 38:5290–5295
Nordlund P, Sjoberg BM, Eklund H (1990) Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 345:593–598
Ono K, Fuma K, Tabata K, Sawada M (2009) Ferritin reporter used for gene expression imaging by magnetic resonance. Biochem Biophys Res Commun 388:589–594
Pereira AS, Tavares P, Lloyd SG, Danger D, Edmondson DE, Theil EC, Huynh BH (1997) Rapid and parallel formation of Fe3+ multimers, including a trimer, during H-type subunit ferritin mineralization. Biochemistry 36:7917–7927
Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH (1998) Direct spectroscopic and kinetic evidence for the involvement of a peroxodiferric intermediate during the ferroxidase reaction in fast ferritin mineralization. Biochemistry 37:9871–9876
Pereira AS, Timóteo CG, Guilherme M, Folgosa F, Naik SG, Duarte AG, Huynh BH, Tavares P (2012) Spectroscopic evidence for and characterization of a trinuclear ferroxidase center in bacterial ferritin from Desulfovibrio vulgaris Hildenborough. J Am Chem Soc 134:10822–10832
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Pfaffen S, Abdulqadir R, Le Brun NE, Murphy MEP (2013) Mechanism of ferrous iron and oxidation by ferritin from a pennate diatom. J Biol Chem 288:14917–14925
Quail MA, Jordan P, Grogan JM, Butt JN, Lutz M, Thomson AJ, Andrews SC, Guest JR (1996) Spectroscopic and voltammetric characterisation of the bacterioferritin-associated ferredoxin of Escherichia coli. Biochem Biophys Res Commun 229:635
Recalcati S, Invernizzi P, Arosio P, Cairo GJ (2008) New functions for an iron storage protein: the role of ferritin in immunity and autoimmunity. J Autoimmun 30:84–89
Ren B, Tibbelin G, Kajino T, Asami O, Ladenstein R (2003) The multi-layered structure of Dps with a novel di-nuclear ferroxidase center. J Mol Biol 329:467–477
Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P (1993) Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature 366:537–543
Salgado EN, Faraone-Mennella J, Tezcan FA (2007) Controlling protein-protein interactions through metal coordination: assembly of a 16-helix bundle protein. J Am Chem Soc 129:13374–13375
Salgado EN, Lewis RA, Faraone-Mennella J, Tezcan FA (2008) Metal-mediated self-assembly of protein superstructures: influence of secondary interactions on protein oligomerization and aggregation. J Am Chem Soc 130:6082–6084
Sana B, Poh CL, Lim S (2012) A manganese-ferritin nanocomposite as an ultrasensitive T2 contrast agent. Chem Commun (Camb) 48:862–864
Sana B, Johnson E, Le Magueres P, Criswell A, Cascio D, Lim SJ (2013) The role of nonconserved residues of Archaeoglobus fulgidus ferritin on its unique structure and biophysical properties. Biol Chem 288:32663–32672
Sirivech S, Frieden E, Osaki S (1974) The release of iron from horse spleen ferritin by reduced flavins. Biochem J 143:311–315
Stefanini S, Vecchini P, Chiancone E (1987) On the mechanism of horse spleen apoferritin assembly: a sedimentation velocity and circular dichroism study. Biochemistry 26:1831–1837
Stillman TJ, Hempstead PD, Artymiuk PJ, Andrews SC, Hudson AJ, Treffry A, Guest JR, Harrison PM (2001) The high-resolution X-ray crystallographic structure of the ferritin (EcFtnA) of Escherichia coli; comparison with human H ferritin (HuHF) and the structures of the Fe(3þ) and Zn(2þ) derivatives. J Mol Biol 307:587–603
Stillman TJ, Connolly PP, Latimer CL, Morland AF, Quail MA, Andrews SC, Treffry A, Guest JR, Artymiuk PJ, Harrison PM (2003) Insight into the effects on metal binding of the systematic substitution of five key glutamate ligands in the ferritin of Escherichia coli. J Biol Chem 278:26275–26286
Swartz L, Kuchinskas M, Li HY, Poulos TL, Lanzilotta WN (2006) Redox-dependent structural changes in the Azotobacter vinelandii bacterioferritin: new insights into the ferroxidase and iron transport mechanism. Biochemistry 45:4421–4428
Takezawa Y, Bockmann P, Sugi N, Wang Z, Abe S, Murakami T, Hikage T, Erker G, Watanabe Y, Kitagawa S, Ueno T (2011) Incorporation of organometallic Ru complexes into apo-ferritin cage. Dalton Trans 40:2190–2195
Tatur J, Hagen WR, Matias PM (2007) Crystal structure of the ferritin from the hyperthermophilic archaeal anaerobe Pyrococcus furiosus. J Biol Inorg Chem 12:615–630
Tatur J, Hagen WR, Heering HA (2009) Voltammetry of Pyrococcus furiosus ferritin: dependence of iron release rate on mediator potential. Dalton Trans 15:2837–2841
Terashima M, Uchida M, Kosuge H, Tsao PS, Young MJ, Conolly SM, Douglas T, McConnell MV (2011) Human ferritin cages for imaging vascular macrophages. Biomaterials 32:1430–1437
Theil EC (2011) Ferritin protein nanocages use ion channels, catalytic sites, and nucleation channels to manage iron/oxygen chemistry. Curr Opin Chem Biol 15:304–311
Theil EC, Behera EK (2013) The chemistry of nature’s iron biominerals in ferritin protein nanocages. In: Ueno T, Watanabe Y (eds) Coordination chemistry in protein cages: principles, design, and applications. Wiley, Hoboken. doi:10.1002/9781118571811
Tosha T, Hasan MR, Theil EC (2008) The ferritin Fe2 site at the diiron catalytic center controls the reaction with O2 in the rapid mineralization pathway. Proc Natl Acad Sci U S A 105:18182–18187
Tosha T, Ng HL, Bhattasali O, Alber T, Theil EC (2010) Moving metal ions through ferritin-protein nanocages from three-fold pores to catalytic sites. J Am Chem Soc 132:14562–14569
Tosha T, Behera RK, Ng HL, Bhattasali O, Alber T, Theil EC (2012) Ferritin protein nanocage ion channels: gating by N-terminal extensions. J Biol Chem 287:13016–13025
Toussaint L, Bertrand L, Hue L, Crichton RR, Declercq JP (2007a) High-resolution X-ray structures of human apoferritin H-chain mutants correlated with their activity and metal-binding sites. J Mol Biol 365:440–452
Toussaint L, Bertrand L, Hue L, Crichton RR, Declercq JP (2007b) High resolution Xray structures of human apoferritin H-chain mutants correlated with their activity and metal-binding sites. J Mol Biol 365:440–452
Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM (1997) Dinuclear center of ferritins: studies of iron binding and oxidation show differences in the two iron sites. Biochemistry 36:432–441
Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM (1998a) The use of zinc(II) to probe iron binding and oxidation by the ferritin (EcFtnA) of Escherichia coli. J Biol Inorg Chem 3:682–688
Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM (1998b) How the presence of three iron binding sites affects the iron storage function of the ferritin (EcFtnA) of Escherichia coli. FEBS Lett 432:213–218
Trikha J, Theil EC, Allewell NM (1995) High resolution crystal structures of amphibian red-cell L ferritin: potential roles for structural plasticity and salvation in function. J Mol Biol 248:949–967
Uchida M, Terashima M, Cunningham CH, Suzuki Y, Willits DA, Willis AF, Yang PC, Tsao PS, McConnell MV, Young MJ, Douglas T (2008) A human ferritin iron oxide nano-composite magnetic resonance contrast agent. Magn Reson Med 60:1073–1081
Uchida M, Kang S, Reichhardt C, Harlen K, Douglas T (2010) The ferritin superfamily: supramolecular templates for materials synthesis. Biochim Biophys Acta 1800:834–845
Wade VJ, Levi S, Arosio P, Treffry A, Harrison PM, Mann S (1991) Influence of site-directed modifications on the formation of iron cores in ferritin. J Mol Biol 221:1443–1452
Wade VJ, Treffry A, Laulhere JP, Bauminger ER, Cleton MI, Mann S, Briat JF, Harrison PM (1993) Structure and composition of ferritin cores from pea seed (Pisum sativum). Biochim Biophys Acta 1161:91–96
Waldo GS, Theil EC (1993) Formation of iron(III)-tyrosinate is the fastest reaction observed in ferritin. Biochemistry 32:13262–13269
Wang Z, Takezawa Y, Aoyagi H, Abe S, Hikage T, Watanabe Y, Kitagawa S, Ueno T (2011) Definite coordination arrangement of organometallic palladium complexes accumulated on the designed interior surface of apo-ferritin. Chem Commun (Camb) 47:170–172
Watt RK (2011) The many faces of the octahedral ferritin protein. BioMetals 24:489–500
Watt GD, Jacobs D, Frankel RB (1988) Redox reactivity of bacterial and mammalian ferritin: is reductant entry into the ferritin interior a necessary step for iron release? Proc Natl Acad Sci U S A 85:7457–7461
Watt GD, Frankel RB, Jacobs D, Huang HQ (1992) Fe2+ and phosphate interactions in bacterial ferritin from Azotobacter vinelandii. Biochemistry 31:5672–5679
Weeratunga SK, Lovell S, Yao H, Battaile KP, Fischer CJ, Gee CE, Rivera M (2010) Structural studies of bacterioferritin B from Pseudomonas aeruginosa suggest a gating mechanism for iron uptake via the ferroxidase center. Biochemistry 49:1160–1175
Wiedenheft B, Mosolf J, Willits D, Yeager M, Dryden KA, Young M, Douglas T (2005) An archaeal antioxidant: characterization of a Dps-like protein from Sulfolobus solfataricus. Proc Natl Acad Sci U S A 102:10551–10556
Wong KK, Colfen H, Whilton NT, Douglas T, Mann S (1999) Synthesis and characterization of hydrophobic ferritin proteins. J Inorg Biochem 76:187–195
Wong SG, Tom-Yew SAL, Lewin A, Le Brun NE, Moore GR, Murphy MEP, Mauk AG (2009) Structural and mechanistic studies of a stabilized subunit dimer variant of Escherichia coli bacterioferritin identify residues required for core formation. J Biol Chem 284:18873–18881
Wong SG, Grigg JC, Le Brun NE, Moore GR, Murphy ME, Mauk AG (2015) The B-type channel is a major route for iron entry into the ferroxidase center and central cavity of bacterioferritin. J Biol Chem 290:3732–3739
Wood JC (2011) Impact of iron assessment by MRI. Hematology Am Soc Hematol Educ Program 2011:443–450
Xing R, Wang X, Zhang C, Zhang Y, Wang Q, Yang Z, Guo Z (2009) Characterization and cellular uptake of platinum anticancer drugs encapsulated in apoferritin. J Inorg Biochem 103:1039–1044
Xu B, Chasteen ND (1991) Iron oxidation chemistry in ferritin. Increasing Fe/O2 stoichiometry during core formation. J Biol Chem 266:19965–19970
Yamashita I, Iwahori K, Kumagai S (2010) Ferritin in the field of nanodevices. Biochim Biophys Acta 1800:846–857
Yang XK, Chen-Barrett Y, Arosio P, Chasteen ND (1998) Reaction paths of iron oxidation and hydrolysis in horse spleen and recombinant human ferritins. Biochemistry 37:9743–9750
Yang X, Le Brun NE, Thomson AJ, Moore CR, Chasteen ND (2000) The iron oxidation and hydrolysis chemistry of Escherichia coli bacterioferritin. Biochemistry 39:4915–4923
Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H, Guo Z (2007a) Encapsulation of platinum anticancer drugs by apoferritin. Chem Commun (Camb) 33:3453–3455
Yang Z, Wang X, Diao H, Zhang J, Li H, Sun H, Guo Z (2007b) Encapsulation of platinum anticancer drugs by apoferritin. Chem Commun (Camb) 33:3453–3455
Zhang Y, Raudah S, Teo H, Teo GW, Fan R, Sun X, Orner BP (2010) Alanine-shaving mutagenesis to determine key interfacial residues governing the assembly of a nano-cage maxi-ferritin. J Biol Chem 285:12078–12086
Zhao G, Bou-Abdallah F, Yang X, Arosio P, Chasteen ND (2001) Is hydrogen peroxide produced during iron(II) oxidation in mammalian apoferritins? Biochemistry 40:10832–10838
Zhao GH, Bou-Abdallah F, Arosio P, Levi S, Janus-Chandler C, Chasteen ND (2003) Multiple pathways for mineral core formation in mammalian apoferritin. The role of hydrogen peroxide. Biochemistry 42:3142–3150
Zhao GH, Su MH, Chasteen ND (2005) mu-1,2-peroxo diferric complex formation in horse spleen ferritin. A mixed H/L-subunit heteropolymer. J Mol Biol 352:467–477
Zheng B, Yamashita I, Uenuma M, Iwahori K, Kobayashi M, Uraoka Y (2010) Site-directed delivery of ferritin-encapsulated gold nanoparticles. Nanotechnology 21:045305
Ziv K, Meir G, Harmelin A, Shimoni E, Klein E, Neeman M (2010) Ferritin as a reporter gene for MRI: chronic liver over expression of H-ferritin during dietary iron supplementation and aging. NMR Biomed 23:523–531
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Yévenes, A. (2017). The Ferritin Superfamily. In: Harris, J., Marles-Wright, J. (eds) Macromolecular Protein Complexes. Subcellular Biochemistry, vol 83. Springer, Cham. https://doi.org/10.1007/978-3-319-46503-6_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-46503-6_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-46501-2
Online ISBN: 978-3-319-46503-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)