
Abstract
A series of oxovanadium complexes with mixed ligands, a tridentate ONO-donor Schiff base ligand [viz., salicylidene anthranilic acid (SAA)], and a bidentate NN ligand [viz., 2,2′-bipyridine (bpy), 1,10-phenanthroline (phen), dipyrido[3,2-d:2′,3′-f]quinoxaline (dpq), dipyrido[3,2-a:2′,3′-c]phenazine (dppz), or 7-methyldipyrido[3,2-a:2′,3′-c]phenazine (dppm)], have been synthesized and characterized by elemental analysis, electrospray ionization mass spectrometry, UV–vis spectroscopy, Fourier transform IR spectroscopy, EPR spectroscopy, and X-ray crystallography. Crystal structures of both complexes, [VIVO(SAA)(bpy)]·0.25bpy and [VIVO(SAA)(phen)]·0.33H2O, reveal that oxovanadium(IV) is coordinated with one nitrogen and two oxygen atoms from the Schiff base and two nitrogen atoms from the bidentate planar ligands, in a distorted octahedral geometry (VO3N3). The oxidation state of V(IV) with d 1 configuration was confirmed by EPR spectroscopy. The speciation of VO–SAA–bpy in aqueous solution was investigated by potentiomtreic pH titrations, and the results revealed that the main species are two ternary complexes at a pH range of 7.0–7.4, and one is the isolated crystalline complex. The complexes have been found to be potent inhibitors against human protein tyrosine phosphatase 1B (PTP1B) (IC50 approximately 30–61 nM), T-cell protein tyrosine phosphatase (TCPTP), and Src homology phosphatase 1 (SHP-1) in vitro. Interestingly, the [VIVO(SAA)(bpy)] complex selectively inhibits PTP1B over the other two phosphatases (approximate ninefold selectivity against SHP-1 and about twofold selectivity against TCPTP). Kinetics assays suggest that the complexes inhibit PTP1B in a competitive and reversible manner. These suggest that the complexes may be promising candidates as novel antidiabetic agents.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Protein tyrosine phosphatases (PTPs) are enzymes that catalyze protein tyrosine dephosphorylation. In humans, over 100 PTPs have been identified that function either as negative or positive modulators in various signal transduction pathways [1, 2]. Dysregulation of PTP activities contributes to the pathogenesis of several human diseases, including diabetes, obesity, cancer, and immune disorders [3–5]. The importance of PTPs in diverse pathogenesis has made them promising targets for drug discovery [6, 7]. Among various members in the PTP superfamily, human PTP 1B (PTP1B) plays a major role in modulating both insulin sensitivity and fuel metabolism and has emerged as an important drug target for the treatment of diabetes and obesity [1, 3, 8]. Recent studies have validated the role of PTP1B as a major contributor to insulin resistance, therefore providing a potential pharmaceutical target for treatment of type II diabetes and obesity [9, 10]. PTP1B inhibitors have been pursued to develop novel antidiabetic drugs [11–16]. A major problem with the current inhibitors is their lack of selectivity towards various PTPs because their active sites are highly conserved, hampering their pharmaceutical development [1]. Thus, potent and specific inhibitors of PTP1B are expected to have high therapeutic value.
Vanadium compounds have long been recognized as phosphatase inhibitors since vanadate has a structure similar to that of phosphate [17]. Numerous studies have demonstrated that vanadium compounds have insulin-enhancing effects and can improve the symptoms of diabetes in a variety of animal models [17–20]. Two inorganic vanadium salts, vanadyl sulfate and sodium metavanadate, have entered phase I clinical trials, but further development is impeded by the poor bioavailability and severe irritation of the digestive tract [21]. Organovanadium complexes were subsequently developed, in which the organic ligands may provide ways for tuning the chemistry and bioactivities of vanadium, thereby minimizing the adverse effects without sacrificing important benefits [22–24]. A pharmacokinetic study performed with carrier-added bis(maltolato)oxovanadium (BMOV) and VOSO4 has demonstrated improved tissue uptake of vanadium from the maltolato-complexed vanadyl compared with its inorganic congener [25]. A BMOV derivative is currently in phase II clinical trials as an antidiabetic drug [26]. The mechanism of action for vanadium’s insulin-sensitizing effects is not well understood but a few studies indicated that association with PTP inhibition is implicated [27–29]. Therefore, how vanadium compounds directly inhibit PTPs and then influence metabolism needs more investigation.
We have been exploring novel vanadium complexes with tridentate Schiff base ligands as PTP1B inhibitors [30]. Here we report the bioactivities in inhibition of three PTPs of five novel mixed-ligand oxovanadium complexes containing a tridentate Schiff base (salicylidene anthranilic acid, SAA) [31] and bidentate polypyridyl ligands (2,2′-bipyridine, bpy; 1,10-phenanthroline, phen; dipyrido[3,2-d:2′,3′-f]quinoxaline, dpq; dipyrido[3,2-a:2′,3′-c]phenazine, dppz; and 7-methyldipyrido[3,2-a:2′,3′-c]phenazine, dppm). The potent inhibitory activity and good selectivity towards PTP1B suggest that these complexes may be promising candidates for novel antidiabetic drug development.
Materials and methods
Reactants
All reagents and solvents were purchased commercially and used without further purification unless specially noted. Double-distilled water was used to prepare buffer solutions. The ligands (SAA, dpq, dppz, and dppm) were synthesized as previously described [32–34].
Physical measurements
Elemental analyses were carried out with a VARI-EL elemental analyzer. IR spectra (4,000–400 cm−1) were recorded using a Shimadzu Fourier transform IR-8300 spectrometer in KBr disks. The electronic spectra were recorded with a Hewlett-Packard HP-8453 Chemstation spectrophotometer in dimethyl sulfoxide (DMSO) solutions. The EPR spectrum was obtained in DMSO solution at 110 K using a Bruker-ER 200-D-SRC spectrometer. Electrospray ionization mass spectra were recorded with a Quattro Micro API instrument (Waters, USA) in methanol aqueous solution (1:9, v/v). The X-ray data were collected using a Bruker SMART APEX 1K CCD diffractometer. Bioactivity assays of the complexes were carried out with a Bio-Rad model 550 microplate reader.
Potentiometric titration
pH-potentiometric titration was performed to study the species formed in aqueous solution. The protonation constant of the ligand (SAA) and the stability constants of the VO–SAA–bpy complex were determined by pH-potentiometric titrations of 40-mL samples. Measurements of the pH values were carried out at 298 ± 0.1 K and at a constant ionic strength of 0.1 M NaCl with a PHS-3TC pH meter with a combined glass electrode. The electrode was calibrated by standard buffer solutions. The titrations were performed with a carbonate-free NaOH solution of known concentration (0.09495 M) by using a microsyringe under a nitrogen atmosphere in a sealed jacketed vessel. The protonation constants of the ligand and the equilibrium constants of the complexes were calculated with the aid of the SUPERQUAD program [35]. The values of pK w of water and pK a of bpy used in the calculations were 13.78 [36] and 6.6 [37], respectively. The overall formation constant of the ternary complex is denoted as the logarithm of β pqrs = [VO p SAA q bpy r H s ]/[VO]p[SAA]q[bpy]r[H]s. The conventional notation has been used: negative indices for H in the formulas indicate either the dissociation of groups which do not deprotonate in the absence of VO(IV) coordination or hydroxo ligands. The following hydroxo species of VO2+ were taken into account in the calculations: [VO(OH)]+ (log β 100-1 = −5.94) and [(VO)2(OH)2]2+ (log β 200-2 = −6.95), with the stability constant calculated from the data of Henry et al. [38] and corrected for the different ionic strengths by use of the Davies equation [39], and [(VO)2(OH)5]− (log β 200-5 = −22.0) [40].
Preparation of oxovanadium(IV) complexes
Five new oxovanadium complexes, each containing a tridentate Schiff base ligand and a bidentate NN ligand, were prepared by a two-step procedure as shown in Scheme 1.

The synthesis of the complexes. SAA salicylidene anthranilic acid, bpy 2,2′-bipyridine, phen 1,10-phenanthroline, dpq dipyrido[3,2-d:2′,3′-f]quinoxaline, dppz dipyrido[3,2-a:2′,3′-c]phenazine, dppm 7-methyldipyrido[3,2-a:2′,3′-c]phenazine
[VIVO(SAA)(bpy)]·H2O
A stirring solution of salicylaldehyde (2 mmol) in 5 mL of methanol was added dropwise to a mixture of anthranilic acid (2 mmol) and KOH (2 mmol), which were dissolved in 10 mL of methanol and stirred at 333 K. After 1 h, a hot aqueous VOSO4 solution (2 mmol) was added. Then the reaction mixture was neutralized by adding aqueous KOH solution. After 2 h of refluxing, a solution of bpy (2 mmol) in 10 mL of methanol was added dropwise to this mixture. The solution on further refluxing for another 3 h gave a reddish-brown precipitate. The solid powder was isolated, washed with distilled water, methanol, and diethyl ether, respectively, and dried in vacuo. Yield: 0.48 g (51%). Anal. data for C24H19N3O5V ([VIVO(SAA)(bpy)]·H2O, 1·H2O)— calcd: C 60.01, H 3.99, N 8.75%; Found: C 60.62, H 4.02, N 8.96%; formula weight (1): 462.35. Electrospray ionization mass spectrometry (ESI-MS): m/z 463.4 [1 + H]+, 485.3 [1 + Na]+. Selected IR data [KBr, ν max (cm−1)]: 3,447br, 1,632s, 1,604vs, 1,581w, 1,530m, 1,461w, 1,439m, 1,344m, 1,188w, 1,151w, 957s, 876m, 761m, 733w, 611w, 419w (br, broad; vs, very strong; s, strong; m, medium; w, weak). When excess bpy (3 mmol) was employed in the last step of the synthesis, reddish-brown crystals of 1·0.25bpy were obtained by slow evaporation of the reaction solution at room temperature for 2 weeks. Suitable dimensional crystals were selected for X-ray single-crystal diffraction.
[VIVO(SAA)(phen)]·H2O
This complex was synthesized following the same general procedures as those described for 1·H2O, with phen in place of bpy. Yield: 0.54 g (53%). Anal. data for C26H19N3O5 V ([VIVO(SAA)(phen)]·H2O, 2·H2O)— calcd: C 61.91, H 3.80, N 8.33%; Found: C 62.39, H 3.99, N 8.16%; formula weight (2): 486.37. ESI-MS: m/z 487.44 [2 + H]+, 509.5 [2 + Na]+. Selected IR data [KBr, ν max (cm−1)]: 3,454br, 1,627s, 1,603vs, 1,578s, 1,531m, 1,457w, 1,422m, 1,359m, 1,182w, 1,144w, 1,032w, 966s, 842m, 777m, 724 m, 614w, 429w. Reddish-brown crystals were obtained by slow evaporation of the reaction solution at room temperature for 2 days.
[VIVO(SAA)(dpq)]·CH3OH
This complex was synthesized following the same general procedures as those described for 1·H2O, with dpq in place of bpy. Yield: 0.52 g (46%). Anal. data for C29H21N5O5V ([VIVO(SAA)(dpq)]·CH3OH, 3·CH3OH)— calcd: C 61.06, H 3.71, N 12.28%; Found: C 60.81, H 3.675, N 12.30%; formula weight (3): 538.41. ESI-MS: m/z 539.3 [3 + H]+, 561.4 [3 + Na]+. Selected IR data [KBr, ν max (cm−1)]: 3,432br, 1,647s, 1,603vs, 1,576s, 1,532m, 1,437w, 1,422m, 1,354m, 1,182w, 1,144w, 1,032w, 970s, 842m, 777m, 724m, 614w, 429w.
[VIVO(SAA)(dppz)]
This complex was synthesized following the same general procedures as those described for 1·H2O, with dppz in place of bpy. Yield: 0.47 g (40%). Anal. data for C32H19N5O4V ([VIVO(SAA)(dppz)], 4)— calcd: C 65.31, H 3.25, N 11.90%; Found: C 64.86, H 3.31, N 11.93%; formula weight (4): 588.47. ESI-MS: m/z 589.4 [4 + H]+, 611.9 [4 + Na]+. Selected IR data [KBr, ν max (cm−1)]: 3,434br, 1,637s, 1,600vs, 1,576m, 1,532s, 1,494m, 1,461m, 1,436m, 1,338m, 1,188w, 1,148w, 972s, 875m, 765m, 736m, 616w, 415w.
[VIVO(SAA)(dppm)]·H2O
This complex was synthesized following the same general procedures as those described for 1·H2O, with dppm in place of bpy. Yield: 0.53 g (43%). Anal. data for C33H23N5O5V ([VIVO(SAA)(dppm)]·H2O, 5·H2O)— calcd: C 63.88, H 3.74, N 11.29%; Found: C 63.36, H 3.741, N 11.27%; formula weight (5): 602.49. ESI-MS: m/z 603.5 [5 + H]+, 626.6 [5 + Na]+. Selected IR data [KBr, ν max (cm−1)]: 3,434br, 1,635s, 1,605vs, 1,578m, 1,532s, 1,495m, 1,461m, 1,438m, 1,358m, 1,183w, 1,148w, 962s, 876w, 756m, 738m, 613w, 419w.
X-ray crystallography
Single crystals of complexes 1·0.25bpy or 2·0.33H 2 O were mounted on glass fibers for data collection. Cell parameters and an orientation matrix for data collection were obtained by least-squares refinement of diffraction data from 1,402 and 2,201 reflections with θ of 2.1–25.0° for 1·0.25bpy and 2·0.33H 2 O using a Bruker SMART APEX 1K CCD automatic diffractometer. Data were collected at 298 K using Mo Kα radiation (λ = 0.71073 Å) and the ω-scan technique, and corrected for Lorentz and polarization effects (SADABS) [41]. The structures were solved by direct methods (SHELXS-97) [42] and subsequent difference Fourier maps and then refined on F 2 by a full-matrix least-squares procedure using anisotropic displacement parameters [43]. Solvent molecule water in 2·0.33H 2 O was found to be disordered with 33% occupancies. After several cycles of refinement, hydrogen atoms attached to carbon atoms were located at their calculated positions (C–H, 0.93Å) and were refined using a riding model. Hydrogen atoms attached to oxygen atoms (water) in 2·0.33H 2 O were located from difference Fourier maps and refined their global U iso value. Molecular graphics are from SHELXTL [44].
Density functional theory calculations and molecular modeling
By using the Gaussian 03 program [45], we performed density functional theory (DFT) structural optimizations on the complexes at the B3LYP/LANL2DZ level [46–48]. The imaginary frequencies and DFT wavefunction instabilities were checked at the same theoretical level. The starting structures of 1 and 2 were derived from the crystal structure and those of other complexes were built on the basis of the crystal structure of complex 2. The molecular modeling (seeing the electronic supplementary information) was performed with an SGI workstation with the insight II software package. The main calculation program was DISCOVER 98 with default settings. An extensible and systematic force field was used with its default parameters.
Expression and purification of recombinant human PTP enzymes
Expression and purification of human PTP1B
Plasmids pET/PTP1B encoding human PTP1B catalytic domain 1–321 and a C-terminal His-tag sequence [30] were transformed into Escherichia coli BL21 (DE3) cells and subsequently plated on Luria–Bertani (LB) plates. A single colony of an overnight culture was inoculated into 1 L fresh liquid LB medium containing 100 mg L−1 ampicillin at 310 K. Expression of PTP1B was induced by isopropyl β-d-thiogalactopyranoside (IPTG) and the His-tagged fusion protein was purified by nickel nitrilotriacetic acid chromatography similarly as described previously for the periplasmic heme-transport protein [49]. The fractions were collected and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The peak fractions containing the pure fusion protein were pooled, and then dialyzed against 3-morpholinopropanesulfonic acid (MOPS) buffer (20 mM MOPS, 0.5 M NaCl, pH 7.4) and stored at 253 K. Protein concentration was determined by the Bradford method using bovine serum albumin as a standard.
Expression and purification of human T-cell PTP and Src homology phosphatase 1
A single clone of E. coli (DE3) cells transformed by the plasmid pT7-ΔTCPTP [50] containing the human T-cell PTP (TCPTP) catalytic domain was inoculated into LB medium containing 100 μg mL−1 ampicillin, 34 μg mL−1 chloramphenicol at 310 K. When the culture reached to an optical density at 600 nm of approximately 0.4, 80 μM IPTG was added to induce the expression of TCPTP at 301 K overnight. The cells were harvested by centrifugation (5,000 rpm, 277 K, 10 min). The pellet was washed with phosphate-buffered saline and resuspended in tris(hydroxymethyl)aminomethane (Tris) buffer [25 mM Tris, 10 mM 2-mercaptoethanol (2-ME), pH 7.5, including 0.1% phenylmethylsulfonyl fluoride and 0.1% phosphoenolpyruvate] and lysed by sonication. The cell debris was removed by centrifugation (12,000 rpm, 277 K, 30 min). The supernatant was directly loaded to an FFQ column and eluted with buffer Q (25 mM Tris, 2 mM 2-ME, pH 7.5) containing 50 mM NaCl. The active peak fraction was loaded onto a SP-Sephadex column and eluted with buffer S (20 mM MeS, 2 mM 2-ME, pH 6.0) containing 150 mM NaCl. The active peak fraction was collected and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis, then concentrated and kept at 253 K. Protein concentration was determined by the Bradford method using bovine serum albumin as a standard.
Src homology phosphatase 1 (SHP-1) constructed as the pT7-ΔSHP-1 expression vector containing human SHP-1 catalytic domain [51] was expressed and purified by almost the same procedure as that for TCPTP. The only difference was that the active peak fraction was eluted from the SP-Sephadex column with buffer S containing 450 mM NaCl.
PTP inhibition assays
PTP activities were measured similarly as described by Montalibet et al. [52] using p-nitrophenol phosphate (pNPP) as the substrate. The assays were performed in 20 mM MOPS buffer, pH 7.2, containing 50 mM NaCl and 2 mM l-glutathione. The complexes were dissolved in DMSO (10−2 M), and diluted to various concentrations, then further diluted 10 times into enzyme–MOPS buffer solutions for activity studies. Inhibition assays were performed in the same buffer on a 96-well plate in 100-μL volumes. The PTPs were diluted to final concentrations of 60, 250, and 200 nM for PTP1B, TCPTP, and SHP-1 respectively. Then 10 μL of the complex with various concentrations was mixed with 83 μL of enzyme solution for 30 min. Then 2 μL of pNPP (0.1 M) substrate was added. After incubation for 30 min at room temperature, the assays were terminated by the addition of 5 μL of 2 M NaOH. A 405 was measured on a microplate reader. IC50 values were obtained by fitting the concentration-dependent inhibition curves using the program Origin. All data points were obtained in triplicate. Solutions of the oxovanadium complexes were all freshly prepared before each experiment.
The inhibiting kinetic analysis was performed according to Eq. 1 [53]:
where V max is the maximum initial velocity, K m is the corresponding constant for substrate, S is the substrate, I is the inhibitor, and K i is the inhibition constant at various substrate concentrations, derived from the slope of the Lineweaver–Burk plots.
Inhibition constants were determined by measuring initial hydrolysis rates at different substrate and inhibitor concentrations. The apparent K i values measured at the various inhibitor concentrations were plotted against concentration of the inhibitor to calculate the K i values [54].
Results and discussion
Description of molecular structures of complexes 1 and 2
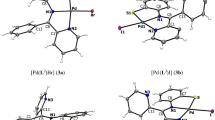
Complexes 1·0.25bpy and 2·0.33H 2 O crystallize in space groups \( P_{{\overline{1} }} \) and \( R_{{\bar{3}}}, \) respectively. Detailed information on the crystal data and structure determination is summarized in Table S1. Selected bond lengths and angles are shown in Table S2. As shown in Fig. 1, the structures of the complexes consist of a monomeric vanadium(IV) species with a VO2+ moiety bonded to a dianionic tridentate Schiff base ligand and an N,N-donor bpy (or phen) base. The complexes have a VIVO3N3 coordination geometry, with the V=O distance of 1.595(3) Å, 1.601(3) Å (1), and 1.589(3) Å (2), typical for a double bond (the “normal” V=O bonds in other oxovanadium complexes are commonly in the range from 1.57 to 1.62 Å) [55–57]. The Schiff base is bound through the phenolate oxygen, the imine nitrogen, and the carboxylate oxygen of the benzoformic acid. The Schiff base exhibits a meridional binding mode, leaving the N,N-donor base bound at the axial–equatorial positions. Owing to the “trans effect” from the oxo ligand, the V–N bond trans to the V=O group is significantly longer [2.322(4) Å, 2.321(4) Å (1), and 2.365(4) Å (2)] than the other V–N bonds (approximately 2.091–2.141 Å), which is well comparable to that in complexes with a similar coordination sphere [58]. The V–O distances involving the Schiff bases are in the range 1.928(4)–1.967(3) Å in 1 [1.957(3) and 1.978(3) Å in 2], typical for single bonds. The lattice water molecule in complex 2 shows intermolecular hydrogen-bonding interactions with the carboxylate oxygen of the Schiff base. The cell packing of both complexes is shown in Figs. S1 and S2.

ORTEP view of 1 (left) and 2 (right) showing atom labeling with 30% probability thermal ellipsoids
DFT calculations were performed for complexes 1 and 2 and the other complexes using the crystal structure of complex 2 as a starting model. The calculated structures are shown in Fig. 2. Selected bond lengths and angles are summarized in Table 1. A comparison of the calculated geometry of complexes 1 and 2 with the structures obtained from single X-ray diffraction indicates a good agreement between computational and experimental results. The DFT calculations suggest that all the complexes adopt a geometry around the vanadyl similar to that in complexes 1 and 2.

The optimized structures of the complexes [VO(SAA)(NN)] (SAA is salicylidene anthranilic acid) at the B3LYP/LANL2DZ level (a NN is 2,2′-bipyridine (bpy), b NN is 1,10-phenanthroline, c NN is dipyrido[3,2-d:2′,3′-f]quinoxaline, d NN is dipyrido[3,2-a:2′,3′-c]phenazine, e NN is 7-methyldipyrido[3,2-a:2′,3′-c]phenazine)
IR spectra
The vanadium complexes 1–5 exhibit a sharp band at approximately 966 cm−1 due to the ν(V=O) mode, indicating that the oxovanadium is in a hexacoordinated environment [54]. The band near 620 cm−1 is assigned to ν(V–N) and the one near 420 cm−1 is assigned to ν(V–O) [59, 60]. The C=N stretching frequencies for the ligands occurring at approximately 1,620 cm−1 are shifted to lower frequencies (1,600–1,605 cm−1) in the complexes, indicating the coordination of the azomethine/ring nitrogen to vanadium [61]. A distinct positive shift of ν(C–O) phenolic oxygen compared with the free Schiff base ligand is observed owing to the formation of the V–O (phenolic oxygen) bond (cf. in the complexes at 1,338–1,359 cm−1 and in the free ligand at 1,234 cm−1) [62]. Furthermore, the peaks at 1,695 and 1,598 cm−1, due to νasym (COO−) and νsym (COO−) of the Schiff base ligand, appear in the complexes at 1,627–1,647 and 1,530–1,578 cm−1. The shifts of these two bands suggest coordination of the carboxylic group oxygen to vanadium in all these complexes [63]. In addition, a broad band appears at approximately 3,400 cm−1, which may be ascribed to hydrogen-bonded ν(O–H) and/or ν(N–H), and may also include ν(C–H).
EPR spectrum
EPR is a useful tool in investigating the nature of bonding in oxovanadium(IV) complexes [64]. The X-band EPR spectrum of complex 2 in 100% DMSO at 110 K is shown in Fig. 3. Similar to the spectra of many other oxovanadium(IV) complexes [62, 65, 66], the spectrum exhibits an axially symmetrical signal of tetravalent vanadium, split into a number of hyperfine lines which originate from the d 1 electron interaction with nuclear spin I = 7/2. The spectrum displays well-resolved 51V (I = 7/2) hyperfine lines (g || > g ⊥) and the parameters (g || = 1.989 and g ⊥ = 1.9541) were obtained from the simulated spectrum. It is clear that the g values obtained are indicative for the presence of vandal ions (VO2+) in a distorted octahedrally coordinated environment [67]. The EPR data are consistent with the solution structure being the same as that characterized in the solid state by X-ray crystallography.

X-band EPR spectrum of complex 2 in dimethyl sulfoxide (DMSO) at 110 K
Electronic absorption spectra
The complexes were insoluble in water and only moderately soluble in methanol, but highly soluble in DMSO. The spectral data of all ligands and the oxovanadium(IV) complexes are displayed in Table S3. The spectra of the ligands show several sharp absorption maxima in the UV region, assignable to π → π* and n → π* transitions. Upon complexation, the π → π* transitions shifted to longer wavelengths, while the n → π* transition merged with an additional broad intense band at approximately 401 nm due to ligand-to-metal charge transfer from the phenolate oxygen to an empty d orbital of vanadium (Fig. 4). In addition, all neutral complexes display two absorptions at 770 and 510 nm with low intensities, assignable to a metal-centered d–d transition. The remaining bands in the UV region are assignable to the intraligand transitions [55, 58, 68]. The stabilities of the complexes in DMSO were also studied at 298 K by UV–vis spectroscopy. No changes were observed for the complexes in DMSO for over 72 h, indicating that the complexes are quite stable in DMSO (Fig. 4).

UV–vis spectra of complexes 1 (a) and 4 (b) recorded at 298 K in DMSO over 72 h with 8-h intervals
ESI-MS analysis
The complexes were further characterized by ESI-MS in methanol aqueous solution (1:9, v/v). Species corresponding to the proton adducts and the sodium adducts were observed for all five complexes (Fig. S3). The composition of the complexes deduced from the elemental analysis was confirmed by the ESI-MS study and the presence of the corresponding parent peak in the positive ion ESI-MS excluded immediate hydrolysis of complexes in methanol aqueous solution [69].
Potentiometric titrations
To obtain information about the complexes formed in aqueous solution, which is relevant to their bioactivities, potentiometric titration was performed for VO2+ cation and two ligands (SAA and bpy). As a preliminary step for studying the VO2+–SAA–bpy system, the protonation constants of SAA were determined and the estimated pK a values were 4.24(14) and 8.90(9). The protonation constants of the bpy ligand and the stability constants of the hydroxo complex of VO2+ were obtained from the literature [37, 38, 40]. Then the titration curves for the binary systems of VO2+–SAA (1:2) and VO2+–bpy (1:3) and the ternary system VO2+–SAA–bpy (1:1:1) were analyzed and the species were suggested according to the data shown in Table 2. On the basis of these data, the species distribution diagram as a function of pH is shown in Fig. 5. The distribution curves suggest that the mixed-ligand oxovanadium species [VO(SAA)(bpy)] and [VO(SAA)(bpy)H-1]− are the predominant species (their percentage is above 90%) in the pH range 7–7.4. The species [VO(SAA)(bpy)] agrees with the structural model from the X-ray structure studies.

Species distribution as a function of pH for the VO2+–SAA–bpy (1:1:1) system (0.9 mM VO2+)
PTP1B inhibition assays
The inhibitory activities of all the [VIVO(SAA)(NN)] complexes on PTP1B were measured using pNPP as the substrate. The enzyme and complexes were preincubated for 30 min prior to the initiation of the enzymatic reaction by the addition of the substrate pNPP. IC50 values were obtained by fitting the dose-dependent inhibition curves based on the means of several replicate experiments. IC50 values of the ligands were determined to be over 1 mM. As shown in Fig. 6, the five oxovanadium complexes demonstrated strong inhibition against PTP1B, with IC50 ranging from 30 to 61 nM. Complex 1 shows the most potent inhibition, with the lowest IC50 value (30 nM). With the increase in size of the polypyridyl ligands, the inhibitory activity gradually decreased, with complex 5 showing the lowest activity (IC50 = 61 nM). It appears that the spatial bulk of the polypyridyl moieties of the complexes weakly diminishes the PTP1B inhibition activities in this series.

Concentration-dependent inhibitions of protein tyrosine phosphatase 1B by complexes 1–5. The inset shows IC50 values
Inhibitions of SHP-1 and TCPTP
As an initial assessment of its specificity, complex 1 was also tested for its inhibitory activities against another two tyrosine phosphatases, SHP-1 and TCPTP, in addition to PTP1B. A comparison of the dose-dependent inhibition of the three PTPs and the IC50 values is shown in Fig. 7. Complex 1 is found to be a potent inhibitor for all three PTPs, with IC50 values in the nanomolar to submicromolar range. Interestingly, the differences in IC50 values suggest that complex 1 appears to be more selective in inhibiting PTP1B than the other two PTPs, with an IC50 value approximately half of that for TCPTP and one ninth of that for SHP-1. Therefore, complex 1 appears to be a PTP1B selective inhibitor, compared with SHP-1 and TCPTP. This interesting selectivity may offer advantages for antidiabetic drug development [15].

Concentration-dependent inhibitions of three tyrosine phosphatases by complex 1. The inset shows IC50 values
Kinetic analysis of PTP1B inhibition
To elucidate the modes of PTP1B inhibition by the oxovanadium complexes, kinetic studies of the inhibitions against PTP1B by complexes 1, 2, and 4 were performed in more detail. Six different concentrations of the substrate pNPP (0.3, 0.5, 1.0, 2.0, 4.0, and 8.0 mM) and five different concentrations of the inhibitors (0, 15, 30, 50, and 80 nM) were used in the steady state kinetics assays. Figure 8 shows the Lineweaver–Burk double-reciprocal plot of the kinetics data in the presence of various of concentrations of complex 1. It appears that the lines converged at an intersection on the y-axis above the x-axis, implying a competitive inhibition mode versus pNPP. Similar phenomena were observed with the other oxovanadium complexes, suggesting that these complexes are likely to be classic competitive inhibitors against PTP1B. A similar inhibition mode has been reported for BMOV [27]. The K i values were determined to be 23 ± 1, 24 ± 1, and 35 ± 3 nM for complexes 1, 2 and 4, respectively.

Lineweaver–Burk plot of 1/ν (min μM−1) versus the reciprocal of the p-nitrophenol phosphate concentration (mM−1) at five fixed concentrations of complex 1
Biological relevance
Type 2 diabetes, characterized by insulin resistance, affects over 200 million people and the epidemic is exploding worldwide [70]. Recently, PTP1B was identified as a key enzyme related to insulin resistance [1–3]. PTP1B inhibition has emerged as an important approach to enhance insulin sensitivity. Various inorganic and organovanadium compounds have been demonstrated to enhance insulin sensitivity in animal models [21]. Organic ligands on vanadium can influence its bioactivity, by affecting either the potency of the phosphatase enzyme inhibition, the selectivity for various phosphatases and kinases, or the bioavailability of the vanadium ion. Careful design of the organic ligands on vanadium could affect their bioactivities significantly, thus making them more suitable for clinical usages in fighting diabetes. Our PTP inhibition data show that the mixed-ligand oxovanadium compounds are highly potent, competitive inhibitors for PTP1B, with IC50 values at the nanomolar level. Moreover, complex 1 demonstrated good selectivity for PTP1B compared with the other two human PTPs. It is worth mentioning that achieving selectivity between PTP1B and TCPTC is extremely challenging [71], because they are approximately 80% homologous in the catalytic domains and share very similar active sites [71]. Our preliminary molecular modeling studies suggest that the oxovanadium complex [VIVO(SAA)(phen)] can be accommodated into the active-site cleft of PTP1B (see the electronic supplementary material). The computer model suggests that the oxovanadium itself can be well fitted into the active-site cleft of PTP1B. In the model, the vanadyl oxygen is close to the sulfur atom of the active-site cysteine (Cys215) (Fig. S4), at a distance of 3.38 Å. Meanwhile, two hydrogen bonds may also be formed between the two oxygens of the carboxyl of the complex and the nitrogens of the arginine residue (Arg221) in the enzyme (Ocarboxyl oxygen–NArg221, 3.13 Å and Ocarbozyl oxygen–NArg221, 3.07 Å) (Fig. S5). These observations suggest that the mixed-ligand oxovanadium complexes may be promising candidates as novel antidiabetic agents or lead compounds for further development.
Conclusion
A series of novel ternary oxovanadium(IV) complexes were synthesized and characterized. The structures of complexes 1 and 2 were determined by X-ray crystal analysis. Potentiometric studies suggest that, in the physiological pH range, VO(IV) can form stable complexes with SAA and bpy. Interestingly, the complexes display potent inhibition of PTP1B, with IC50 values in low nanomolar range. Kinetic analysis indicates that these complexes are reversible competitive inhibitors of PTP1B. Moreover, complex 1 appears to be a PTP1B selective inhibitor, compared with SHP-1 and TCPTP. These observations make the oxovanadium(IV) complexes promising candidates for novel antidiabetic drug development.
Abbreviations
- BMOV:
-
Bis(maltolato)oxovanadium
- bpy:
-
2,2′-Bipyridine
- dppm:
-
7-Methyldipyrido[3,2-a:2′,3′-c]phenazine
- DFT:
-
Density functional theory
- DMSO:
-
Dimethyl sulfoxide
- dppz:
-
Dipyrido[3,2-a:2′,3′-c]phenazine
- dpq:
-
Dipyrido[3,2-d:2′,3′-f]quinoxaline
- ESI-MS:
-
Electrospray ionization mass spectrometry
- IPTG:
-
Isopropyl β-d-thiogalactopyranoside
- LB:
-
Luria–Bertani
- 2-ME:
-
2-Mercaptoethanol
- MOPS:
-
3-Morpholinopropanesulfonic acid
- pNPP:
-
p-Nitrophenol phosphate
- phen:
-
1,10-Phenanthroline
- PTP:
-
Protein tyrosine phosphatase
- PTP1B:
-
Protein tyrosine phosphatase 1B
- SAA:
-
Salicylidene anthranilic acid
- SHP-1:
-
Src homology phosphatase 1
- TCPTP:
-
T-cell protein tyrosine phosphatase
- Tris:
-
Tris(hydroxymethyl)aminomethane
References
Zhang S, Zhang ZY (2007) Drug Discov Today 12:373–381
Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T (2004) Cell 117:699–711
Zhang ZY (2001) Curr Opin Chem Biol 5:416–423
Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Moller NPH (2004) FASEB J 18:8–30
Arena S, Benvenuti S, Bardelli A (2005) Cell Mol Life Sci 62:2092–2099
Combs AP, Yue EW, Bower M, Ala PJ, Wayland B, Douty B, Takvorian A, Polam P, Wasserman Z, Zhu WY, Crawley ML, Pruitt J, Sparks R, Glass B, Modi D, McLaughlin E, Bostrom L, Li M, Galya L, Blom K, Hillman M, Gonneville L, Reid BG, Wei M, Becker-Pasha M, Klabe R, Huber R, Li YL, Hollis G, Burn TC, Wynn R, Liu P, Metcalf B (2005) J Med Chem 48:6544–6548
Dewang PM, Hsu NM, Peng SZ, Li WR (2005) Curr Med Chem 12:1–22
Zhang ZY, Lee SY (2003) Expert Opin Investig Drug 12:223–233
Goldstein BJ (2001) Curr Drug Targets Immune Endocr Metabol Disord 1:175–265
Seale AP, de Jesus LA, Kim SY, Choi YH, Lim HB, Hwang CS, Kim YS (2005) Biotechnol Lett 27:221–225
Xie LP, Lee SY, Andersen JN, Waters S, Shen K, Guo XL, Moller NPH, Olefsky JM, Lawrence DS, Zhang ZY (2003) Biochemistry 42:12792–12804
Yuen VG, Caravan P, Gelmini L, Glover N, McNeill JH, Setyawati IA, Zhou Y, Orvig C (1997) J Inorg Biochem 68:109–116
Shrestha S, Bhattarai BR, Lee KH, Cho H (2007) Bioorg Med Chem 15:6535–6548
Sparks RB, Polam P, Zhu W, Crawley ML, Takvorian A, McLaughlin E, Wei M, Ala PJ, Gonneville L, Taylor N, Li Y, Wynn R, Burn TC, Liu PCC, Combs AP (2007) Bioorg Med Chem Lett 17:736–740
Maccari R, Paoli P, Ottana R, Jacomelli M, Ciurleo R, Manao G, Steindl T, Langer T, Vigorita MG, Camici G (2007) Bioorg Med Chem 15:5137–5149
Winter CL, Lange JS, Davis MG, Gerwe GS, Downs TR, Peters KG, Kasibhatla B (2005) Exp Biol Med 230:207–216
Crans DC, Smee JJ, Gaidamauskas E, Yang LQ (2004) Chem Rev 104:849–902
Rehder D (2003) Inorg Chem Commun 6:604–617
Shechter Y, Goldwaser I, Mironchik M, Fridkin M, Gefel D (2003) Coord Chem Rev 237:3–11
Li M, Ding WJ, Baruah B, Crans DC (2008) J Inorg Biochem 102:1846–1853
Thompson KH, Orvig C (2006) J Inorg Biochem 100:1925–1935
Noblia P, Baran EJ, Otero L, Draper P, Cerecetto H, Gonzalez M, Piro OE, Castellano EE, Inohara T, Adachi Y, Sakurai H, Gambino D (2004) Eur J Inorg Chem 322–328
Crans DC, Smee JJ (2004) Comp Coord Chem II 4:175–239
Ghosh T, Bhattacharya S, Das A, Mukherjee G, Drew MGB (2005) Inorg Chim Acta 358:989–996
Thompson KH, Orvig C (2000) Dalton Trans 2885–2892
Mukherjee R, Donnay E.G, Radomski MA, Miller C, Redfern DA, Gericke A, Damron DS, Brasch NE (2008) Chem Commun 3783–3785
Peters KG, Davis MG, Howard BW, Pokross M, Rastogi V, Diven C, Greis KD, Eby-Wilkens E, Maier M, Evdokimov A, Soper S, Genbauffe F (2003) J Inorg Biochem 96:321–330
Huyer G, Liu S, Kelly J, Moffat J, Payette P, Kennedy B, Tsaprailis G, Gresser MJ, Ramachandran C (1997) J Biol Chem 272:843–851
Nxumalo F, Glover NR, Tracey AS (1998) J Biol Inorg Chem 3:534–542
Lu LP, Zhu ML, Guo ML (unpublished work)
Patel RN, Singh N, Gundla VLN (2006) Polyhedron 25:3312–3318
Dickeson JE, Summers LA (1970) Aus J Chem 23:1023–1027
Amouyal E, Homsi A, Chambron JC, Sauvage JP (1990) J Chem Soc Dalton Trans 1841–1845
Clague MJ, Keder NL, Butler A (1993) Inorg Chem 32:4757–4761
Gans P, Sabatini A, Vacca A (1985) J Chem Soc Dalton Trans 1195–1200
Moghimi A, Alizadeh R, Shokrollahi A, Aghabozorg H, Shamsipur M, Shockravi A (2003) Inorg Chem 42:1616–1624
Martell AE, Chaberek S Jr, Courtney RC, Westerback S, Hyytiainen H (1957) J Am Chem Soc 79:3036–3041
Henry RP, Mitchell PCH, Prue JE (1973) J Chem Soc Dalton Trans 1156–1159
Davis CW (1938) J Chem Soc 2093–2098
Chruscinska EL, Sanna D, Garribba E, Micera G (2008) Dalton Trans 4903–4916
Sheldrick GM (1996) Correction software. University of Göttingen, Göttingen
Sheldrick GM (1997) Program for the solution of crystal structure. University of Göttingen, Göttingen
Sheldrick GM (1997) Program for the refinement of crystal structure. University of Göttingen, Göttingen
Sheldrick GM (1999) SHELXTL/PC, version 5.1. Bruker AXS, Madison
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven JT, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, revision E.01. Gaussian, Wallingford
Hay PJ, Wadt WR (1985) J Chem Phys 82:270–283
Hay PJ, Wadt WR (1985) J Chem Phys 82:284–298
Hay PJ, Wadt WR (1985) J Chem Phys 82:299–310
Tong Y, Guo ML (2007) J Biol Inorg Chem 12:735–750
Zhu ZC, Sun M, Zhang XY, Liu KX, Shi DL, Li JD, Su JQ, Xu YC, Fu XQ (2007) Chem Res Chin Univ 23:289–296
Li WN, Zhuang Y, Li H, Sun Y, Fu Y, Wu XX, Zhao ZZ, Fu X (2008) Chem Res Chin Univ 24:592–596
Montalibet J, Skorey KI, Kennedy BP (2005) Methods 35:2–8
Ember B, Kamenecka T, LoGrasso P (2008) Biochemistry 47:3076–3084
Mondal S, Rath SP, Rajak KK, Chakravorty A (1998) Inorg Chem 37:1713–1719
Maurya MR, Kumar A, Ebel M, Rehder D (2006) Inorg Chem 45:5924–5937
Root CA, Hoeschele JD, Cornman CR, Kampf JW, Pecoraro VL (1993) Inorg Chem 32:3855–3861
Amin SS, Cryer K, Zhang B, Dutta SK, Eaton SS, Anderson OP, Miller SM, Reul BA, Brichard SM, Crans DC (2000) Inorg Chem 39:406–416
Sasmal PK, Patra AK, Nethaji M, Chakravarty AR (2007) Inorg Chem 46:11112–11121
Zamian JR, Dockal ER, Castellano G, Oliva G (1995) Polyhedron 14:2411–2418
Marques APD, Dockal ER, Skrobot FC, Rosa ILV (2007) Inorg Chem Commun 10:255–261
Westland AD, Tarafder MTH (1981) Inorg Chem 20:3992–3995
Bhattacharya S, Ghosh T (2002) Transit Metal Chem 27:89–94
Soliman AA, Mohamed GG (2004) Thermochim Acta 421:151–159
Raman N, Raja SJ, Joseph J, Raja JD (2007) Russ J Coord Chem 33:7–11
Mathieu M, Van Der Voort P, Weckhuysen BM, Rao RR, Catana G, Schoonheydt RA, Vansant EF (2001) J Phys Chem B 105:3393–3399
Pessoa JC, Cavaco I, Correia I, Costa D, Henriques RT, Gillard RD (2000) Inorg Chim Acta 305:7–13
Ghosh T, Bandyopadhyay C, Bhattacharya S, Mukherjee G (2004) Transit Metal Chem 29:444–450
Dutta SK, Kumar SB, Bhattacharyya S, Tiekink ERT, Chaudhury M (1997) Inorg Chem 36:4954–4960
Stepanenko IN, Krokhin AA, John RO, Roller A, Arion VB, Jakupec MA, Keppler BK (2008) Inorg Chem 47:7338–7347
Wild S, Roglic G, Green A, Sicree R, King H (2004) Diabetes Care 27:1047–1053
Lee S, Wang Q (2006) Med Res Rev 27:553–573
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (grant no. 20471033), the Natural Science Foundation of Shanxi Province (grant no. 20051013), the Overseas Returned Scholar Foundation of Shanxi Province of China in 2006 and 2008, and University of Massachusetts Dartmouth, MA, USA.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
775_2009_496_MOESM1_ESM.pdf
Supplementary material 1 Supporting Information Available: X-ray crystallographic data in CIF format of complex 1 (CCDC708872) and 2 (CCDC707122). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk. Detailed information on the crystal data and structure determination (Table S1), Selected bond lengths and angles for complexes 1 and 2 (Table S2), Unit cell packing diagrams for complexes 1 and 2 (Figs. S1 and S2), UV–vis data for the ligands and the complexes (Table S3), ESI-MS data (Figs. S3, A-E) and molecular modeling of the interactions of complex 2 at the active site of PTP1B (Figs. S4 and S5). (PDF 619 kb)
Rights and permissions
About this article
Cite this article
Yuan, C., Lu, L., Gao, X. et al. Ternary oxovanadium(IV) complexes of ONO-donor Schiff base and polypyridyl derivatives as protein tyrosine phosphatase inhibitors: synthesis, characterization, and biological activities. J Biol Inorg Chem 14, 841–851 (2009). https://doi.org/10.1007/s00775-009-0496-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-009-0496-6