
Abstract
Four oxovanadium(IV) complexes, namely [VO(desa-met)(phen)]·MeOH·2H2O (1) (desa-met = Schiff base derived from 4-(diethylamino)salicylaldehyde and dl-methionine, phen = 1,10-phenanthroline), [VO(o-van-met) (phen)]·MeOH·CH2Cl2·3H2O (2) (o-van-met = Schiff base derived from o-vanillin and dl-methionine), [VO(dtbs-napa)(phen)]·2H2O (3) (dtbs-napa = Schiff base derived from 3,5-di-tert-butyl salicylaldehyde and 3-(1-naphthyl)-l-alanine) and [VO(hyna-napa)(phen)]·1.5H2O (4) (hyna-napa = Schiff base derived from 2-hydroxy-1-naphthaldehyde and 3-(1-naphthyl)-l-alanine), were synthesized and characterized by IR, HRMS, UV–vis spectra, molar conductance and single-crystal X-ray diffraction (XRD). X-ray structural analysis showed that the V(IV) atoms in all four complexes are six-coordinated in a distorted octahedral environment. In the crystals of complexes 1 and 2, π–π stacking interactions together with hydrogen bonds connect the molecular units into 2D networks. Meanwhile, CH–π stacking interactions are observed between the aromatic rings in the crystals of 1 and 4, while the π–π stacking interactions between aromatic rings in the crystals of 2 and 3 are arranged with a face-to-face mode. The in vitro anticancer activities of these complexes against A-549 and HeGp2 cells were tested by MTT assay.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cisplatin was the first platinum-based complex to be used as an effective anticancer drug [1, 2]. Cisplatin and related Pt(II) complexes exhibit remarkable anticancer activities; however, they suffer from problems related to severe side effects in normal tissue, nephrotoxicity, drug resistance and cost per patient. These drawbacks limit the use and efficiency of cisplatin and related Pt(II) complexes in cancer therapy [3–5]. These considerations have stimulated the search for new non-platinum anticancer agents that possess reduced side effects, lower resistance, lower cost and greater efficacy toward a wider range of cancers [6, 7]. Hence, in the past few decades, transition metal complexes have been extensively studied as anticancer and antibacterial agents [8, 9]. In particular, oxovanadium salts are less expensive than K2PtCl4, RuCl3 or VCl3 and are therefore attractive for vanadium-based research and development of inexpensive drugs. Current research suggests that transformed cells maintain an abnormal redox homeostasis that helps them to obtain a high level of genetic instability, which is conducive to cancer progression [10–12]. Vanadium’s intrinsic redox activity can generate reactive oxygen species (ROS). This enhanced level of ROS can disorganize the redox homeostasis in neoplastic cells and impart redox stress to the cell, either by cleaving DNA or by promoting mitochondrial membrane permeabilization [13]. Moreover, a growing body of evidence reveals that vanadium complexes can act as potent anticancer agents [14]. Although inorganic vanadium salts have relatively high toxicity and poor biological activity, the complexation of vanadium with organic ligands can minimize adverse effects while enhancing its benefits [15]. Therefore, it is important to synthesize novel oxovanadium complexes and investigate their medical applications.
Organic ligands containing Schiff bases, considered privileged structures, have attracted considerable attention due to their facile synthesis and variable coordination behavior toward metal ions, as well as their wide applications in various fields (e.g., as chemosensors [16], catalysts [17], anticancer agents [18, 19], adsorbents [20] and bactericidal agents [21, 22]). Efforts have been made to synthesize and characterize amino acid Schiff base complexes of transition metals [23, 24]. Recently, our research group, and others, reported some vanadium complexes with anticancer activities [15, 19]. In this work, four new amino acid Schiff base oxovanadium(V) complexes have been synthesized and characterized (Fig. 1). Moreover, their anticancer activities have been investigated against A-549 and HeGp2 cell lines by the MTT assay (laboratory test and standard colorimetric assay).

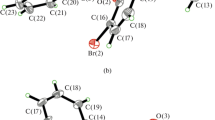
The molecular structure of oxovanadium(IV) complexes 1, 2, 3 and 4
Experimental
Materials and instrumentation
All chemicals and reagents obtained from commercial sources were of AR grade and used without further purification. HRMS (ESI–MS) spectra were recorded using a time-of-flight Micromass LCT Premier XE spectrometer. Elemental (C, H, N) analyses were obtained with a Vario EL III elemental analyzer. IR spectra were recorded on KBr pellets, with a Vary FT-IR 1000 spectrophotometer in the range 400–4000 cm−1. Molar conductivity measurements were obtained using a QCJD-2010 (China) conductivity meter.
Synthesis of complexes 1 and 2
Complexes 1 and 2 were prepared by a one-pot synthetic procedure in which a mixture of dl-methionine (0.14 g, 1.0 mmol) and KOH (0.06 g, 1.0 mmol) in 15 mL hot methanol–water (v:v = 1:1) was added to a solution of 4-(diethylamino)-salicylaldehyde (0.21 g, 1.0 mmol) or o-vanillin (0.15 g, 1.0 mmol) in ethanol (5 mL). The mixture was refluxed for 2 h, followed by addition of a solution of vanadyl sulfate (0.16 g, 1.0 mmol) in water (5 mL). The reaction was continued for another 1 h, followed by addition of a solution of 1,10-phenanthroline (0.20 g, 1.0 mmol) in 5 mL methanol. The resulting solution, after further refluxing for 1 h, gave a red precipitate. The solid was isolated by filtration, washed with small amounts of water, methanol and diethyl ether and dried. Single crystals of 1 and 2 suitable for X-ray diffraction were obtained over one week via slow evaporation of MeOH-CH2Cl2 (v:v = 1:1) solutions at room temperature.
Complex 1, [VO(desa-met)(phen)]·MeOH·2H2O: Yield: 72.8 %. Anal. Calcd for C28H30N4O4SV·CH3OH·2H2O: C 54.6, H 6.0, N 8.8. Found: C 54.8, H 6.0, N 8.6. HRMS (ESI–MS) calculated for C28H30N4O4SVNa (M + Na)+: 593.1359, found m/z: 593.1310. FTIR: 3469(s), 3416(s), 3241(w), 2975(m), 2904(w), 1657(s), 1616(s), 1598(m), 1515(w), 1399(m), 953(m), 879(w), 728(w), 601(w), 484(w). UV–vis in DMSO:H2O (v:v = 1:1), 2.0 × 10−6 mol/L, (λ max/nm) 265, 354. Λ m = 4.7 Ω−1 mol−1 cm2 in DMSO: H2O (v:v) = 1:1 at 25 °C.
Complex 2, [VO(o-van-met)(phen)]·MeOH·CH2Cl2·3H2O: Yield: 75.3 %. Anal.Calcd for C25H23N3O5SV·CH3OH·CH2Cl2·3H2O: C 46.4, H 5.0, N 6.0. Found: C 46.5, H 5.1, N 6.1. HRMS (ESI–MS) calculated for C25H23N3O5SVNa (M + Na)+: 551.0668, found m/z: 551.0696. FTIR: 3469(s), 3415(s), 3231(m), 3018(w), 2920(w), 1638(s), 1617(s), 1535(w), 1516(w), 1400(m), 960(m), 850(m), 726(m), 618(w), 476(w). UV–vis in DMSO:H2O (v:v = 1:1), 2.0 × 10−6 mol/L, (λ max/nm) 262, 390, 410. Λ m = 5.7 Ω−1 mol−1 cm2 in DMSO:H2O (v:v = 1:1) at 25 °C.
Synthesis of complexes 3 and 4
To a mixture of 3-(1-Naphthyl)-l-alanine (0.22 g, 1.0 mmol) and potassium hydroxide (0.06 g, 1.0 mmol) in 15 mL hot methanol–water (v:v = 1:1) was added to a solution of 3,5-di-tert-butyl salicylaldehyde (0.23 g, 1.0 mmol) or 2-hydroxy-1-naphthaldehyde (0.17 g, 1.0 mmol) in ethanol (5 mL). The mixture was refluxed for 2 h, followed by addition of a solution of vanadyl sulfate (0.16 g, 1.0 mmol) in water (5 mL). The resulting solution was then heated to reflux for another 1 h, followed by the addition of a solution of 1,10-phenanthroline (0.20 g, 1.0 mmol)] in methanol (5 mL). The resulting solution was refluxed for a further refluxing 1 h, giving a yellow precipitate. The solid was isolated, washed with small amounts of water, methanol and diethyl ether and dried. Single crystals of complexes 3 and 4 suitable for X-ray diffraction were obtained over one week via slow evaporation of MeOH-CH2Cl2 (v:v = 1:1) solutions at room temperature.
The data of 3, [VO(3,5-ditbsal-3-1-Naph-l-ala)(phen)]·2H2O: Yield: 76.3 %. Anal. Calcd for C40H39N3O4V·2H2O: C 67.4, H 6.1, N 5.9. Found: C 67.5, H 6.1, N 5.8. HRMS (ESI–MS) calculated for C40H39N3O4VNa (M + Na)+: 699.2278, found m/z: 699.2235. FTIR: 3468(s), 3421(s), 1636.7(s), 1617(s), 1544(w), 1466(w), 1400 (m), 958(m), 848(w), 727(w). UV–vis in DMSO:H2O (v:v = 1:1), 2.0 × 10−6 mol/L, (λ max/nm) 264, 369. Λ m = 3.1 Ω−1 mol−1 cm2 in DMSO:H2O (v:v = 1:1) at 25 °C.
The data of 4, [VO(2-hyd-1-nade-3-1-Naph-l-ala)(phen)]·1.5H2O: Yield: 78.6 %. Anal.Calcd for C36H25N3O4V·1.5H2O: C 67.4, H 4.4, N 6.5. Found: C 67.3, H 4.3, N 6.4. HRMS (ESI–MS) calculated for C36H25N3O4VNa (M + Na)+: 637.1182, found m/z: 637.1173. FTIR: 3550(s), 3473(s), 3416(s), 3236(w), 1637(s), 1617(s), 1540(w), 1510(w), 1456(w), 1399(m), 1095(w), 963(m), 727(w), 617(w), 477(w). UV–vis in DMSO:H2O (v:v = 1:1), 2.0 × 10−6 mol/L, (λ max/nm) 265, 355. Λ m = 6.4 Ω−1 mol−1 cm2 in DMSO: H2O (v:v) = 1:1 at 25 °C.
Crystal Structure Determination
Single-crystal XRD data were collected using a Rigaku diffractometer with a mercury charge-coupled device area detector (Mo Kα; λ = 0.71073 Å) at room temperature. Empirical absorption corrections were applied to the data using the Crystal Clear program [25]. The structure was solved by direct methods and refined by the full-matrix least-squares method on F 2 using the SHELXTL-97 program [26]. Metal atoms were located from E-maps, and other non-hydrogen atoms were located in successive difference Fourier syntheses. All non-hydrogen atoms were refined anisotropically. The organic hydrogen atoms were positioned geometrically, and those in water molecules were located using the difference Fourier method and refined freely.
PLATON/SQUEEZE was used to remove the heavily disordered water molecules. Crystallographic data and other pertinent information for complexes 1–4 are summarized in Table 1. Selected bond distances and angles are listed in Tables S2–S5†. Bond lengths and angles of hydrogen bonds are listed in Tables S6† and S7†.
In vitro anticancer activities
A549 cells (human lung carcinoma cell line) and HepG2 cells (human hepatoma cell line) were purchased from ATCC. A549 cells were grown in DMEM/HIGH GLUCOSE(1X) (Dulbecco’s modified eagle’s medium), and HepG2 cells were maintained in modified Roswell Park Memorial Institute 1640 (RPMI-1640) which were supplemented with 10 % fetal bovine serum and 1 % penicillin streptomycin solution in a humidified atmosphere of 5 % CO2, 95 % air at 37 °C. The passage number range for both cell lines was maintained between 10 and 20. The cells were cultured in 25 cm2 cell culture flasks. For experimental purposes, A549 and HepG2 cells were cultured in 96-well plates, respectively (5 × 104 cells/mL, 100 mL/well). Cells were allowed to attach for 24 h before treatment with the test complexes. A stock solution of each complex was prepared in DMSO and filtered with Minisart filters (0.45 μm). Each stock solution was diluted with serum-free medium to different concentrations (0–120 μM). Cell monolayers were washed with PBS, and the test complexes were added within a range of concentrations from 0 to 120 μM for 24 h. The concentration range for the complexes 1–4 and the exposure times have been selected based on preliminary studies performed in our laboratory.
The MTT assay is based on the protocol originally described by Mossmann [19]. The assay was optimized for the cell lines used in this experiment. Briefly, at the end of the incubation time, cells were incubated for 4 h with 0.5 mg/mL of MTT, dissolved in serum-free medium (DMEM or RPMI-1640 for A459 and HepG2 cells, respectively). Washing with PBS (100 μL) was followed by the addition of DMSO (200 μL) and gentle shaking for 10 min. The absorbance was recorded at 490 nm using Multiskan Spectrum. The cell viability ratio was calculated by the following formula:
Cytotoxicity is expressed as the concentration of the complex inhibiting cell growth by 50 % (IC50 value see Table 1).
Results and Discussion
Synthesis and Characterization
Complexes 1–4 have been prepared in high yields from a one-pot synthetic procedure in which vanadyl sulfate is reacted with the required dianionic α-amino acid Schiff base ligand and 1,10-phenanthroline in aqueous methanol.
The complexes were characterized by their IR spectra, UV–vis spectra and HRMS (ESI–MS) mass spectral data (Figures S1–S9†). All four complexes have small molar conductivity values in 50 % DMSO-H2O at 25 °C, indicating that they are non-electrolytes [19, 27, 28].
Complexes 1–4 gave satisfactory C, H and N analyses. The HRMS (ESI–MS) mass spectra of complexes 1–4 are shown in Figures S1–S4†. The parent ion peaks are observed at m/z 593.1310 for 1, 551.0668 for 2, 699.2235 for 3 and 637.1173 for 4. In addition, the dominant features in the HRMS spectra, namely cationic fragment peaks at m/z 203.0588 to 203.0608, can be associated with the fragment [phen + Na+]+, indicating that all four complexes possess a phen ligand.
The FTIR spectra of complexes 1–4 are shown in Figures S5–S8†. Bands are observed at 953 cm−1 for 1, 960 cm−1 for 2, 958 cm−1 for 3 and 963 cm−1 for 4, assigned to the υ(V=O) stretching vibration, which is typical for oxovanadium complexes [27, 28]. The very sharp absorptions at 1657–1636 cm−1 are characteristic of the imine group, υ(C=N), in the complexes [27, 28]. Weak bands at 848–728 cm−1 are attributed to the ring-stretching frequencies [υ(C=C) and υ(C=N)] of 1,10-phenanthroline [27]. Two moderate absorptions at 1616–1617 cm−1 and 1399–1400 cm−1 can be assigned to the asymmetric and symmetric stretching vibrations of the \( \upsilon_{{\left( {{\text{CO}}_{2}^{ - } } \right)}} \) group [29], respectively. The frequency separation (Δυ) is greater than 200 cm−1, suggesting unidentate bonding for the carboxyl group [30]. Weak peaks in the low wavenumber region 400–650 cm−1 may be attributed to υ(V–O) and υ(V–N) bonds in the complexes [28].
The UV–visible spectra of 1–4 are shown in Figure. S9†. All four complexes display a ligand-centered band at ca. 260 nm, assignable to the π → π* transition. 1,10-Phenanthroline with its respective quinoxaline moiety exhibits an additional band near 350 nm, assignable to the n → π* transition [28].
Description of crystal structures
The molecular structures of complexes 1–4 with the atom numbering schemes are shown in Figs. 2, 3, 4 and 5 and selected bond lengths and angles are given in Table S2–S4†, respectively. The complexes are similar in each of the structures, with the general formulation [VO2L(Phen)]. All four complexes are mononuclear with distorted octahedral geometry. The V(IV) centers are six-coordinated, by two N atoms from one phen ligand, one O atom from the VO2+ moiety, two O atoms and another N atom from the tridentate Schiff base ligand (L). The terminal vanadyl V=O distances are 1.600(14) Å for 1, 1.588(2) Å for 2, 1.579(5) Å for 3 and 1.573(6) Å for 4, which are all in the 1.56–1.76 Å range reported for other vanadyl complexes [19, 31, 32]. The V1–O1 (phenolate oxygen) bond distances are 1.952(13) for 1, 2.00(2) for 2, 1.908(5) for 3 and 1.920(5) for 4, which are as expected from related VO Schiff base complexes [27, 32]. The V1–O2 (carboxylate oxygen) bond lengths at 1.978(14) for 1, 1.951(2) for 2, 2.021(5) for 3 and 1.992(5) for 4 are in the range found for other VO Schiff base complexes [33, 34]. The V(1)–N(3) amine bond lengths are 2.0333(15) Å for 1, 2.048(3) Å for 2, 2.0105(56) for 3 and 2.1294(68) Å for 4. The V(1)–N(2)(phen) bond trans to the V=O group is significantly longer [2.3603(16)–2.3955(70) Å] than the other V(1)–N(1)(phen) distances [2.1294(68)–2.1587 (60) Å]; the respective average length of 2.38 and 2.14 Å is similar to the values reported in the literature [27, 32–34]. In addition, the asymmetric unit contains one solvate methanol molecule in structures 1 and 2, and one solvate dichloromethane molecule is also found in structure 2 (Fig. 3).

Molecular structure of 1 with thermal ellipsoids at 45 % probability (the solvent molecules were omitted)

Molecular structure of 2 with thermal ellipsoids at 45 % probability (the solvent molecules were omitted)

Molecular structure of 3 with thermal ellipsoids at 45 % probability

Molecular structure of 4 with thermal ellipsoids at 45 % probability
In the supramolecular structures of complexes 1 and 4, CH–π stacking interactions between the phen and Schiff base ligands are observed. The H atom of one aromatic ring and another aromatic ring centroid are arranged in an edge-to-face mode with the distances of 2.7447(6) and 2.6998(4) Å, respectively (Figs. 6, 7). In addition, some classic hydrogen bonds are observed in structure 1. These intramolecular hydrogen bonds expand the structure into a supramolecular structure. The π–π stacking interactions together with the hydrogen bonds in the structure form a 2D network (Fig. 8). The lengths and angles of the hydrogen bonds of 1 are given in Table S5†.

The CH–π stacking interactions in 1

The CH–π stacking interactions in 4

The 2D network of 1 by the hydrogen bonds interaction
The supramolecular structures of complexes 2 and 3 show π–π stacking interactions between the aromatic rings. Two aromatic rings are arranged in a face-to-face mode with center of mass distance of 3.4055(35) and 3.4993(43) Å, respectively (Figs. 9, 10).

The π–π stacking interactions in 2

The π–π stacking interactions in 3
Hydrogen bonds between the O–H groups of the solvent MeOH molecules and the uncoordinated carboxylate O atoms extend the structure of 2 into a 2D layer architecture, along with π–π stacking interactions (Fig. 11). The bond lengths and angles of the hydrogen bonds of 2 are given in Table S6†.

The 2D network of 2 by the hydrogen bonds interaction
In vitro anticancer activities
The anticancer properties of these complexes and of cis-Pt against A-549 and HeGp2 cell lines were tested by the MTT assay. Cytotoxicity is expressed as the concentration of the complex inhibiting cell growth by 50 % (Table 2).
Complexes 2–4 were found to have moderate anticancer activities toward A549 (human lung carcinoma cell line) and HepG2 (human hepatoma cell line), with IC50 valves of 27.0–46.3 μmol L−1. Although the activities of these complexes are lower than the controls of cis-Pt and VO(acac)2 [32], they may serve as a starting point for further research and development.
Conclusion
Four new amino acid Schiff base oxovanadium(IV) complexes have been synthesized and characterized. The crystal structures of all four complexes show that their V(IV) atoms have distorted octahedral coordination geometries. Complexes 2–4 have moderate anticancer activities toward human lung carcinoma and human hepatoma cell lines.
Supplementary Material
CCDC 1438533, 1438534, 1438535 and 1438536 contain the supplementary crystallographic data for 1, 2, 3 and 4, respectively. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK. Fax: +44 1223 336 033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Wilson JJ, Lippard SJ (2012) J Med Chem 55:5326–5336
Jany T, Moreth A, Gruschka C, Sischka A, Spiering A, Dieding M, Wang Y, Samo SH, Stammler A, Bogge H, Mollard GF, Anselmetti D, Glaser T (2015) Inorg Chem 54:2679–2690
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G (2012) Oncogene 31:1869–1883
Pabla N, Dong Z (2008) Kidney Int 73:994–1007
Miller RP, Tadagavadi RK, Ramesh G, Reeves WB (2010) Toxins 2:2490–2518
Aher SB, Muskawar PN, Thenmozhi K, Bhagat PR (2014) Eur J Med Chem 81:408–419
Wanninger S, Lorenz V, Subhanb A, Edelmann FT (2015) Chem Soc Rev 44:4986–5002
Almodares Z, Lucas SJ, Crossley BD, Basri AM, Pask CM, Hebden AJ, Phillips RM, McGowan PC (2014) Inorg Chem 53:727–736
Yuan ZL, Shen XM, Huang JD (2015) RSC Adv 5:10521–10528
Yuan ZL, Shen XM, Huang JD, Gang W (2015) J Incl Phenom Macrocycl Chem 82:135–143
Trachootham D, Alexandre J, Huang P (2009) Nat Rev Drug Discov 8:579–591
Barrio DA, Etcheverry SB (2010) Curr Med Chem 17:3632–3642
Strianese M, Basile A, Mazzone A, Morello S, Turco MC, Pellecchia C (2013) J Cell Physiol 228:2202–2209
Evangelou AM (2002) Crit Rev Oncol Hematol 42:249–265
Ebrahimipour SY, Sheikhshoaie I, Kautz AC, Ameri M (2015) Polyhedron 93:99–105
Yang J, Yuan ZL, Yu GQ, He SL, Hu QH, Wu Q, Jiang B, Wei G (2015) J. Fluorescence 26:43–51
Xu YJ, Keiichi K, Motomu K, Masakatsu S, Shigeki M (2014) J Am Chem Soc 136:9190–9194
Chow MJ, Licona C, Wong DYQ, Pastorin G, Gaiddon C, Ang WH (2014) J Med Chem 57:6043–6059
Cao YP, Yi QL, Liu HM, Li HX, Zuo JL, Yuan ZL (2015) Chin J Synth Chem 23:1124–1129
Sapana K, Ghanshyam SC (2014) ACS Appl Mater Interfaces 6:5908–5917
Yuan ZL, Yang QWuXB, Hu QH, Zhang MQ (2011) Chin J Org Chem 31:1698–1702
Hina Z, Anis A, Asad UK, Tahir AK (2015) J Mol Struct 1097:129–135
Zeinab MS, Amini Z, Davar MB, Notash B (2013) Polyhedron 53:76–82
Ozaki Y, Kawashima T, Abe-Yoshizumi R, Kandori H (2014) Biochemistry 53:6032–6040
CrystalClear (2000) Version 1.36, Molecular Structure Corp and Rigaku Corp., The Woodlands
Sheldrick GM SHELXS 97, program for crystal structure solution. University of Göttingen, Göttingen
Sasmal PK, Patra AK, Nethaji M, Chakravarty AR (2007) Inorg Chem 46:11112–111121
Sheng GH, Han X, You ZL, Li HH, Zhu HL (2014) J Coord Chem 67:1760–1770
Vančo J, Trávníek Marek J Z, Račanská E, Muselík J, Vajlenová O (2008) J Inorg Biochem 102:595–605
Neelakantan MA, Rusalraj F, Dharmaraja J, Johnsonraja S, Jeyakumar T, Pillai MS (2008) Spectrochim Acta A 71:1599–1609
Bian L, Li LZ, Zhang QF, Dong JF, Xu T, Li JH, Kong JM (2012) Trans Met Chem 37:783–790
Habala L, Bartel C, Giester G, Jakupec MA, Keppler BK, Rompel A (2015) J Inorg Biochem 147:147–152
Cao YP, Yi QL, Liu HM, Li HX, Zuo JL, Yuan ZL (2015) J Zunyi Med Univ 38(6):584–590
Balaji B, Somyajit K, Banik B, Nagaraju G, Chakravarty AR (2013) Inorg Chim Acta 400:142–150
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81360471), the International Cooperation Project of Guizhou Province (No. [2012]7036), Science and the ‘Chunhui’ plan project of Ministry of Education (No. Z2014089) and the National Project of Training Programs of Innovation and Entrepreneurship for Undergraduates, China (No. 201510661007).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cao, Y., Yi, C., Liu, H. et al. Syntheses, crystal structures and in vitro anticancer activities of oxovanadium(IV) complexes of amino acid Schiff base and 1,10-phenanthroline ligands. Transit Met Chem 41, 531–538 (2016). https://doi.org/10.1007/s11243-016-0049-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-016-0049-0