
Abstract
We describe the clinical and genetic findings in pedigree with a novel mutation in the calcium sensing receptor (CaSR) gene and the unusual coexistence of primary hyperparathyroidism (HPT) and familial hypocalciuric hypercalcaemia (FHH) and its clinical management. The occurrence of both FHH and primary HPT in the same patient has been described rarely. Our pedigree has a novel mutation in the CaSR gene. Parathyroidectomy led to a reduction, but not normalization of the calcium levels in the patient identified as having HPT. The coexistence of HPT and FHH was considered in this patient as her calcium and PTH levels were rising with time. Surgical resection of her parathyroid adenoma resulted in reduction of her calcium levels to above normal and significant reduction in her symptoms of fatigue and low mood.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The calcium-sensing receptor (CaSR) is a plasma membrane-bound protein that is predominantly expressed in the parathyroids and the kidneys, where it allows regulation of parathyroid hormone secretion and renal tubular calcium reabsorption appropriate to the prevailing extracellular calcium concentration [1]. CaSR is also expressed in the gut, pancreas and bone. The human CaSR gene is located on chromosome 3q13-q21 [2]. It is a large glycoprotein that belongs to family C or family 3 of the superfamily of G protein-coupled receptors [3]. It is 1078 amino acids long and has a large extracellular domain, a seven-transmembrane-spanning region, and an intracellular tail [4].
Following cloning of the CaSR from bovine parathyroid in 1993, inactivating mutations of the receptor were shown to be the cause of two inherited conditions of hypercalcaemia- familial hypocalciuric hypercalcaemia (FHH) and neonatal severe hyperparathyroidism (NSHPT) [5]. These are Mendelian disorders and are characterised by an altered response to calcium with an increase in the calcium ion-dependent set-point for parathyroid hormone release from the parathyroid cell [2].
In FHH there is typically a modest elevation of the serum calcium concentration (serum calcium within 10 % of the upper limit of normal), relative hypocalciuria and PTH levels that are not suppressed by the hypercalaemia and are inappropriately normal [1, 4]. NSHPT is the homozygous phenotype of FHH. It is a potentially lethal disease resulting in skeletal demineralisation and multiple fractures [6].
Primary HPT is due to a parathyroid adenoma, diffuse hyperplasia or rarely carcinoma. It can also occur as part of different familial endocrinopathies including multiple endocrine neoplasia (MEN) 1, MEN 2A and isolated familial hyperparathyroidism [2]. Hypercalcaemia due to inappropriate secretion of parathyroid hormone is characteristic and target organ complications are associated [7].
The coexistence of FHH and primary HPT has been described in isolated reports, however, it is unclear if this is coincidental or if a causative link between the two conditions exists [7–9]. We present a kindred with FHH due to a novel genetic mutation. In addition, the index case was diagnosed with and treated for primary HPT due to a functioning parathyroid adenoma.
Case report
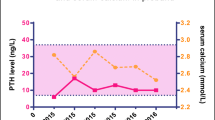
The index case (I1) (Fig. 1a) presented age 63 with an incidental finding of hypercalcaemia. Her past medical history was significant for cholelithiasis, chronic constipation (no gastrointestinal pathology identified) and nephrolithiasis diagnosed at age 33 which was managed conservatively. FHH was suspected based on biochemical workup (Table 1) and genetic testing identified a novel point mutation in the 4th exon of the CaSR gene (CGG to CCG) leading to a change (R220P) in the large extra cellular domain of the protein. She was followed over a period of eight years and biochemical monitoring took place. She was noted to have a gradual increase in her serum calcium and parathyroid hormone levels during this time and reported fatigue and low mood (Table 1).

a Pedigree of the kindrid. The proband is indicated by an arrow. Males and females with normocalcaemia and negative for the CaSR gene mutation are donated by open squares and open circles, respectively. Males and females with hypercalcaemia and positive for the gene mutuation are denoted by filled squares and filled circles, respectively. b Parathyroid (Sesta-MIBI) scintigraphy of the index case. The arrow points to a right lower parathyroid adenoma
Due to her symptomatology and a rise in her calcium and PTH levels over time, parathyroid (Sesta-MIBI) scintigraphy was completed and this revealed a right lower parathyroid adenoma (Fig. 1b). A DEXA scan revealed osteopenia at the lumbar spine (T score −1.6) and neck of femur (T score −1.9). Vitamin D level was 31 nmol/L (reference range >25 nmol/L), magnesium 1.07 mmol/L (normal range 0.70–1.00 mmol/L) and urinary calcium/creatinine ratio was 0.013. The patient proceeded to surgery and a right lower parathyroid adenoma was identified and removed. Histological analysis confirmed the diagnosis of parathyroid adenoma. The rationale for the diagnosis of an adenoma is that the lesion is encapsulated with a rim of uninvolved parathyroid tissue and this is supported by its weight of 618 mg and adipocyte reduction (Fig. 2). There were no surgical complications and post-operatively, the patient reported an improvement in her mood and energy levels. Her laboratory findings 1 year post operatively are outlined in Table 1 and her urinary calcium/creatinine ratio was 0.008.

Right lower parathyroid gland, weight 618 mg. Thinly encapsulated parathyroid tissue is evident with a rim of uninvolved gland. In many areas there is diffuse growth of predominantly oxyphil cells with scattered adipocytes (H&E, ×2)
The pedigree is illustrated (Fig. 1a) and their laboratory findings are listed in Table 1. Family screening revealed that a 32 year-old daughter of the index case (II2) complained of chronic constipation and was diagnosed with mild depression. Her laboratory investigations were consistent with FHH and are outlined in Table 1. Her parathyroid (Sesta-MIBI) scintigraphy was normal.
A second daughter (II4) aged 39 years suffered constipation, nausea, and mild abdominal discomfort. She had a normal parathyroid (Sesta-MIBI) scintigraphy and DEXA scan. Her son (III5) aged 11 years had a past history significant for chronic constipation that was managed conservatively. Both were diagnosed with FHH and remained asymptomatic following advice to increase fluid intake.
Finally, a son of the index case (II3) attended clinic aged 36 years. He was asymptomatic on presentation but reported having abdominal cramps and chronic constipation as a teenager. His biochemical findings were also consistent with FHH.
The other family members were all normocalcaemic and asymptomatic. Genetic testing on affected family members confirmed the same mutation present in the index case. To our knowledge, this is the first kindred of FHH described with this novel mutation.
Discussion
Familial hypocalciuric hypercalcaemia which accounts for 1–2 % cases of hypercalcaemia is generally managed conservatively as parathyroidectomy typically does not lead to normalization of calcium levels or resolution of symptoms [7]. Pathological examination of parathyroid glands resected from patients with FHH has demonstrated glandular enlargement associated with a spectrum of histologic findings but most commonly mild parathyroid hyperplasia. However, FHH is not typically associated with adenoma formation [10].
Primary HPT comprises the majority of cases of hypercalcaemia among ambulatory patients and parathyroidectomy is frequently recommended for patients who develop target organ effects [7, 11].
Distinction between primary HPT and FHH is based on history (for example hypercalcaemia in family members including young children) and biochemical findings such as urinary calcium levels. Approximately 40 % of patients with hyperparathyroidism have hypercalciuria but in FHH, calcium excretion is typically below 5 mmol/day with clearance calcium/clearance creatinine <0.01 [12, 13]. The demonstration of a mutation in the CaSR gene consolidated the diagnosis of FHH in this case. Asymptomatic members of the family had normal serum calcium and were negative for the gene mutation affecting the CaSR. This finding suggests pathogenicity of this mutation. Additional evidence to support pathogenicity of this variant includes reports in the literature demonstrating that other amino acid substitutions at this residue segregate with FHH and calcium response curves indicating that residue 220 is important for ligand-receptor interaction [4].
Of particular interest in this kindred is the presence of a functioning parathyroid adenoma in the proband which is a rare event. In recent years, however, our knowledge of hypercalcaemic disorders has expanded. A German series highlighted the coexistence of HPT and FHH in 4 out of 139 patients with hypercalcaemia and suggested a pathogenic role of CaSR mutations in HPT [14]. A Swedish pedigree with hypercalcaemia was found to have an inactivating point mutation in the cytoplasmic tail of the CaSR along with elevated PTH, magnesium and urinary calcium levels. Subtotal parathyroidectomy revealed parathyroid gland hyperplasia/adenoma and corrected the biochemical disorder in seven of nine individuals. These findings are atypical of both FHH and primary HPT [15].
One case of a patient with FHH and a parathyroid adenoma was reported in 2002 [8] and two cases in 2009 [9, 16]. Earlier this year two further patients were reported and in all cases surgical resection confirmed the diagnosis of parathyroid adenoma and resulted in a lowering of the serum calcium levels [7]. As is true in our case, no causative link between the CaSR mutation and the parathyroid adenoma has been identified in these patients, however, a pathogenic role of the CaSR mutation cannot be excluded. Decreased expression or function of the CaSR gene leads to an altered number of normally functioning cell surface receptors, which in turn may play a role in the proliferation of parathyroid cells. Therefore, one can speculate that total or partial loss of receptor function may stimulate the development of parathyroid adenomas [14]. Close monitoring of affected family members over time will provide more information in this regard.
Conclusion
The coexistence of HPT and FHH was considered in this patient as her calcium and PTH levels were rising with time. Surgical resection of her parathyroid adenoma resulted in reduction of her calcium levels to above normal and significant reduction in her symptoms of fatigue and low mood.
References
Thakker RV (2004) Diseases associated with the extracellular calcium-sensing receptor. Cell Calcium 35:275–282
Carmliet G, Cromphat SV, Daci E, Maes C, Bouillon R (2003) Disorders of calcium homeostasis. Best Pract Res Clin Endocrinol Metab 17:529–546
Brown EM (1983) Four-parameter model of the sigmoidal relationship between parathyroid hormone release and extracellular calcium concentration in normal and abnormal parathyroid tissue. J Clin Endocrinol Metab 56:572–581
D’Souza-Li L, Yang B, Canaff L, Vai M, Hanley DA, Bastepe M, Salisbury SR, Brown EM, Cole DEC, Hendy GN (2002) Identification and functional characterisation of novel calcium-sensing receptor mutations in familial hypocalciuric hypercalcaemia and autosomal dominant hypocalcaemia. J Clin Endocrinol Metab 87:1309–1318
Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG (1993) Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75:1297–1303
Bouschet T, Martin S, Henley JM (2008) Regulation of calcium-sensing-receptor trafficking and cell-surface expression by GPCRs and RAMPs. Trends Pharmacol Sci 29:633–639
Eldeiry LS, Ruan DT, Brown EM, Gaglia JL, Garber JR (2012) Primary hyperparathyroidism and FHH: relationships and Clinical implications. Endocr Pract 18:412–417
Burski K, Torjussen B, Paulsen AQ, Boman H, Bollerslev J (2002) Familial hypocalciuric hypercalcemia: coincidence or causality? J Clin Endocrinol Metab 87:1015–1016
Brachet C, Boros E, Tenoutasse S, Lissens W, Andry G, Martin P, Bergmann P, Heinrichs C (2009) Association of parathyroid adenoma and familial hypocalciuric hypercalcaemia in a teenager. Eur J Endocrinol 161:207–210
Thorgeirsson U, Costa J, Marx SJ (1981) The parathyroid glands in familial hypocalciuric hypercalcaemia. Hum Pathol 12:229–237
Bilezikan JP, Khan AA, Potts JT Jr, on behalf of the Third International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism (2009) Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Third International Workshop. J Clin Endocrinol Metab 94:335–339
Silverberg SJ, Shane E, Jacobs TP, Siris ES, Gartenberg F, Seldin D, Clemens TL, Bilezikian JP (1990) Nephrolithiasis and bone involvement in primary hyperparathyroidism. Am J Med 89:327–334
Marx SJ, Stock JL, Attie MF, Downs RW Jr, Gardner DG, Brown EM, Speigel AM, Doppman JL, Brennan MF (1980) Familial hypocalciuric hypercalcaemia: recognition among patients referred after unsuccessful parathyroid exploration. Ann Intern Med 92:351–356
Frank-Raue K, Leidig-Bruckner G, Haag C, Schulze E, Lorenz A, Schmitz-Winnenthal H, Raue R (2011) Inactivating calcium-sensing receptor mutations in patients with primary hyperparathyroidism. Clin Endocrinol 75:50–55
Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P, Trivedi S, Hellman P, Brown EM, Dahl N, Rastad J (2000) Familial hypercalcaemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab 85:2042–2047
Yabuta T, Miyauchi A, Inoue H, Yoshida H, Hirokawa M, Amino N (2009) A patient with primary hyperparathyroidism associated with familial hypocalciuric hypercalcaemia induced by a novel germline CaSR mutation. Asian J Surg 32:118–122
Acknowledgments
We are indebted to Professor A. Lienhardt and Dr. Corinne Magdelaine from the Laboratoire de Biochemie et Genetique de l’Hopital Universitaire Dupytren de Limoges (France) and Dr. Anneke Seller from the Oxford Genetics Laboratories (England) who carried out the sequencing of the CaSR gene.
Conflict of interest
All authors have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Egan, A.M., Ryan, J., Aziz, M.A. et al. Primary hyperparathyroidism in a patient with familial hypocalciuric hypercalcaemia due to a novel mutation in the calcium-sensing receptor gene. J Bone Miner Metab 31, 477–480 (2013). https://doi.org/10.1007/s00774-012-0399-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00774-012-0399-4