
Abstract
Familial hypocalciuric hypercalcemia (FHH) is a benign autosomal dominant condition characterized by lifelong asymptomatic hypercalcemia. FHH is typically caused by a heterozygous inactivating mutation of the calcium-sensing receptor (CaSR) and characterized by moderate hypercalcemia, inappropriately normal or elevated serum parathyroid hormone (PTH), and relative hypocalciuria (FeCa < 2%) with histologically normal parathyroid glands. FHH should be distinguished from primary hyperparathyroidism so that unnecessary parathyroid surgery is avoided. We report a case that presented with asymptomatic, familial hypercalcemia but low PTH and normal (non-low) urinary calcium excretion found to be secondary to a novel pathogenic inactivating mutation of the CaSR gene. We present an asymptomatic 54-year-old Malaysian woman with incidentally discovered hypercalcemia, intermittent hypophosphatemia, and FeCa > 2%. PTH levels were repeatedly below the mean of the reference range (on two separate assays) and sometimes even below the lower reference limit. Two siblings, one niece, and her son had hypercalcemia without nephrolithiasis. Cinacalcet, used as a PTH-suppression test, normalized serum total and ionized calcium after 7 days of cinacalcet 30 mg BID, confirming her hypercalcemia was PTH-mediated. Given her family history, genetic testing was pursued and discovered a novel pathogenic mutation of the CaSR gene confirming the diagnosis of FHH type 1. Our case represents an atypical presentation of FHH1 with low PTH and FeCa > 2%. This contributes to the expanding clinical and biochemical spectrum of CaSR inactivating mutations and presents an innovative approach to evaluating biochemically uncertain familial hypercalcemia with cinacalcet before pursuing expensive genetic analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Familial hypocalciuric hypercalcemia (FHH) is a benign autosomal dominant condition that is characterized by lifelong, non-progressive, and asymptomatic hypercalcemia [1,2,3]. FHH type 1 is typically caused by a heterozygous inactivating mutation of the gene coding for the calcium-sensing receptor (CaSR), which regulates parathyroid hormone (PTH) secretion and renal calcium excretion [1, 3, 4]. At present, more than 139 mutations in the CaSR gene causing FHH1 have been reported [5]. Less commonly, FHH may be due to mutations of the G protein subunit alpha11 gene (GNA11, FHH type 2), or in the adaptor-related protein complex 2 gene (AP2S1, FHH type 3) [6]. The classic biochemical abnormalities found in FHH include moderate hypercalcemia, inappropriately normal or elevated serum PTH, and relatively low urinary calcium with histologically normal parathyroid glands [1, 4, 7]. FHH should be distinguished from primary hyperparathyroidism (PHPT), so that unnecessary parathyroid surgery is avoided [8].
We report a case of asymptomatic, familial hypercalcemia but low to normal levels of measured serum PTH and normal (non-low) urinary calcium excretion. A logical and innovative approach was used in the evaluation of our patient’s familial hypercalcemia and atypical biochemistry (i.e., suppressed PTH and normocalciuria) using a short course of cinacalcet as a diagnostic calcium-normalization test to demonstrate her hypercalcemia was PTH/CaSR mediated. Genetic sequencing discovered a presumptive novel pathogenic inactivating mutation of the CaSR gene, thereby confirming the diagnosis of FHH type 1.
Case presentation
A 54-year-old woman from Malaysia was assessed in a general endocrinology clinic for incidentally discovered, asymptomatic hypercalcemia. Informed written consent for publication was given. She had no history of nephrolithiasis, gastroesophageal reflux or constipation, osteoporosis, fractures, renal insufficiency, or any psychiatric complaints. Her past medical history was significant for post-traumatic chronic myofascial pain syndrome for which she used analgesics (i.e., cyclobenzaprine, ibuprofen, gabapentin) as needed. She denied supplemental calcium intake beyond a single daily multivitamin and the use of any medications that would influence calcium metabolism including hydrochlorothiazide or lithium. Her family history was reported to include unexplained hypercalcemia without nephrolithiasis in two siblings, one niece, and her son.
On examination, she appeared well and was non-obese (BMI 21.4 kg/m2). Head and neck examination revealed no band keratopathy. There was no evidence of ectopic or subcutaneous calcification. Her proximal muscle strength and reflexes were normal.
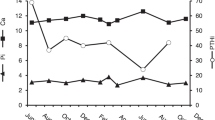
Her bloodwork over a 14-month period is shown in Tables 1 and 2 and Fig. 1 (supplementary material). She had calcium levels that ranged between 2.56 and 2.86 mmol/L (reference 2.10–2.55 mmol/L) with normal albumin, borderline low phosphate between 0.71 and 0.95 mmol/L (reference 0.80–1.50 mmol/L), 24 h urine calcium 6.12 mmol/day (reference 2.50–7.50 mmol/day), and urinary fractional excretion of calcium (FeCa) 2.13%. Serum PTH levels were repeatedly well below the mean of the reference range and occasionally even below the 5th percentile (mean 11 ng/L, range 6–17 ng/L, reference 7–37 ng/L). Remaining bloodwork including serum protein electrophoresis, calcitriol, calcidiol, estimated glomerular filtration rate, thyroid-stimulating hormone, parathyroid hormone-related peptide levels, and bone mineral density were normal. Table 2 shows representative biochemical data from 17 contemporaneously diagnosed patients at our center with primary hyperparathyroidism due to surgically proven adenoma, along with biochemical results from a contemporaneously diagnosed subject from a large and previously characterized FHH family.

Longitudinal biochemical observations in proband; shaded area represents the normal reference range on the PTH assay
Given her clear family history of hypercalcemia but unconvincing biochemistry for FHH, we used a short course of cinacalcet as a diagnostic tool, to distinguish whether her hypercalcemia was CaSR-mediated and judiciously direct further investigations. Baseline total and ionized calcium, phosphate, magnesium, PTH, and spot FeCa levels were obtained before administering cinacalcet 30 mg PO twice daily for 7 days. On the seventh day of therapy, the above measures were repeated and demonstrated clear evidence of calcium normalization and further suppression of PTH, confirming her hypercalcemia was mediated by the CaSR (Table 3). Importantly, she reported no new clinical symptoms during this short treatment course.
Once her hypercalcemia was established to be CaSR-mediated, genetic sequencing was pursued and revealed a presumptive novel pathogenic mutation in her CaSR gene (heterozygous c.1377 + 1G > T variant), confirming the diagnosis of FHH type 1. This variant is predicted to abolish the splice donor site of exon 4 and has not been reported in ExAC or the literature. Most of the proband’s reportedly affected family members live in Malaysia thus clinical and genetic assessment was not possible. However, the proband’s 27-year-old son has recently been found to have mild hypercalcemia and subsequent testing revealed a similar biochemical profile (calcium 2.59 mmol/L (reference range 2.10–2.55 mmol/L), PTH 13 and 20 (reference range 17–37 ng/L), 24 h urine calcium 5.38 mmol/day, and FeCa 1.99%) and the same novel CaSR gene mutation (heterozygous c.1377 + 1G > T variant).
Discussion
FHH is a benign form of PTH-mediated hypercalcemia caused by a heterozygous inactivating mutation of CaSR [1, 4] leading to compensatory hyperparathyroidism, hypercalcemia with relative hypocalciuria [3, 9]. CaSR is expressed in the parathyroid glands and kidneys, where it maintains calcium homeostasis by regulating PTH secretion and renal tubular calcium reabsorption [3]. Inactivating germline CaSR mutations result in an altered calcium ion-dependent set point for PTH release from the parathyroid chief cells and a rightward shift of the PTH-serum calcium dose-response curve [8]. This altered set point explains the inappropriately normal or high serum PTH seen in FHH [10]. This same mutated CaSR in the renal tubular cells leads to increased tubular reabsorption of calcium and a relatively low renal calcium excretion (i.e., relative hypocalciuria FeCa < 1%) [9].
The CaSR is a member of the G protein-coupled receptor family expressed uniformly in all tissues participating in systemic mineral metabolism [11, 12]. It is comprised of 1078 amino acids and contains a 612 amino acid extracellular domain (ECD) essential for co-translational processing, receptor dimerization, binding ligands, and transmitting activation signals through the seven transmembrane domains (TMD’s), made up of residues 613–862, and the intracellular domain (ICD), made up of residues 863–1078 [3, 8]. The majority of FHH1-associated mutations are located at the ECD; however, they can occur at the TMD, ICD, or as a consequence of defective trafficking to the plasma membrane resulting in reduced cell surface receptor expression [8].
Our findings confirm our patient’s atypical biochemistry (suppressed PTH, lack of hypocalciuria) are still a result of a heterozygous CaSR mutation (c.1377 + 1G > T) predicted to abolish the splice site of exon 4, producing a novel genotype of FHH1. Abolishment of the exon 4 splice donor site would result in a truncated CaSR extracellular domain in the cysteine-rich domain region of the CaSR [13]. The deletion of a portion of this domain results in loss of cysteine at position 541 and 545 and is hypothesized to result in compromised function of the CaSR due to the role of the cysteine-rich region in signal transmission from the Venus flytrap (VFT) domain to the seven-transmembrane domain [14, 15]. The impact of alteration of the cysteine-rich domains on the CaSR function is variable and has been shown to tolerate a significant degree of amino acid substitution [14, 15] such that the full impact of exon 4 deletion is unclear. Potential explanations for her atypical biochemical presentation include a variable FHH1 phenotype caused by the underlying CaSR splice donor site mutation leading to loss of a portion of the cysteine-rich extracellular domain or an altered PTH gene resulting in suboptimal detection with some PTH assays [16].
Several authors have reported patients with FHH can exhibit variability in serum calcium and PTH values depending on the underlying CaSR mutation [3, 17, 18]. Variability in serum calcium levels amongst 15 individual families with FHH was described as far back as 1981 by Marx et al. However, CaSR gene sequencing was not performed in these cases to correlate the different genotypes with their biochemical presentation [19]. More recently, a Danish population study evaluated 66 FHH patients with 11 different CaSR mutations and demonstrated marked differences in the biochemical FHH phenotype, with significant inter-mutation variation in both plasma calcium and PTH values [12]. However, none of the reported cases had serum PTH levels below the reference range as seen with our case. Perhaps most strikingly, Aida et al. describe a Japanese woman who inherited two copies of a CaSR missense mutation from her related heterozygous parents resulting in a homozygous gene mutation, classically associated with severe neonatal hyperparathyroidism. However, her genotype only caused a modest defect in her CaSR function and resulted in a mild clinical and biochemical phenotype that was not detected until later in life [20].
Limitations to the interpretation of our patient’s biochemistry include the impact dietary calcium may have on urinary calcium excretion, as well as the method of urinary calcium measurement. However, in healthy individuals, a higher intake of calcium is typically only associated with a small increase in urinary calcium [21]. Additionally, studies based on healthy volunteers ingesting varying degrees of calcium supplementation, have reported a “plateau effect” of calcium intake on urinary calcium excretion [22, 23]. We used the calcium creatinine clearance ratio, also known as FeCa, and spot urine calcium measurements to quantify our patient’s hypercalciuria. FeCa, as an index of renal calcium excretion, has performed equally well when compared to 24-h renal calcium excretion and 24-h renal calcium/creatinine excretion ratio as a discriminative test for patients with FHH and PHPT [24]. In contrast, when searching for overt hypercalcemia, spot urine calcium measurements should not be used interchangeably with 24-h values due to their low sensitivity [25].
In cases of uncertainty regarding the role of PTH and the CaSR in hypercalcemic patients, the CaSR agonist cinacalcet may be of use as a diagnostic tool. Cinacalcet is a calcimimetic that increases the sensitivity of the CaSR in both the parathyroid gland and kidney to calcium. In patients with hyperparathyroidism or CaSR-mediated hypercalcemia, cinacalcet should lower PTH and increase renal calcium excretion thus lowering serum calcium [26]. In our case, we hypothesized the administration of cinacalcet would result in a decrease in serum calcium if our patient’s hypercalcemia was indeed PTH/CaSR-mediated. Her biochemical response to this test (calcium normalization and further suppression of PTH) confirmed our theory. With the concomitant knowledge of the family history of hypercalcemia, it was then logical to pursue genetic testing for FHH.
The use of cinacalcet as a diagnostic tool for routine PHPT has been described previously by Cailleux et al. [26]. The authors found 60 mg of cinacalcet provided similar PTH lowering results as the potentially more dangerous calcium loading test [26]. Although their patient population did not include atypical or FHH patients, this study provides evidence that the use of cinacalcet is safe, effective, and has many potential applications as a diagnostic test where PTH levels are equivocal in the face of hypercalcemia.
Conclusion
We report a case of FHH1 that presented with familial hypercalcemia, suppressed serum PTH, and normocalciuria caused by a novel inactivating CaSR mutation. The underlying mechanism of her atypical biochemistry reminds clinicians to maintain a high index of suspicion for FHH in cases of familial hypercalcemia. Our case contributes to the growing knowledge of the expanding clinical and biochemical spectrum of CaSR inactivating mutations and affords an innovative approach to evaluating biochemically uncertain familial hypercalcemia with cinacalcet before pursuing expensive genetic analysis.
Abbreviations
- FHH:
-
familial hypocalciuric hypercalcemia
- CaSR:
-
calcium-sensing receptor
- PTH:
-
parathyroid hormone
- PHPT:
-
primary hyperparathyroidism
- GNA11:
-
G protein subunit alpha11 gene
- AP2S1:
-
adaptor-related protein complex 2 gene
- BMI:
-
body mass index
- FeCa :
-
fractional excretion of calcium
- ExAC:
-
exome aggregation consortium
- ECD:
-
extracellular domain
- TMD:
-
transmembrane domains
- ICD:
-
intracellular domain
- FHH1:
-
familial hypocalciuric hypercalcemia type 1
- VFT:
-
Venus flytrap
References
Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P et al (2000) Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab 85(5):2042–2047
Thakker RV (2004) Diseases associated with the extracellular calcium-sensing receptor. Cell Calcium 35(3):275–282
Christensen SE, Nissen PH, Vestergaard P, Mosekilde L (2011) Familial hypocalciuric hypercalcaemia: a review. Curr Opin Endocrinol Diabetes Obes 18(6):359–370
Mastromatteo E, Lamacchia O, Campo MR, Conserva A, Baorda F, Cinque L et al (2014) A novel mutation in calcium-sensing receptor gene associated to hypercalcemia and hypercalciuria. BMC Endocr Disord 14:81
CASRdb Calcium Sensing Receptor Locus Mutation Database [Internet]. Available from: http://www.casrdb.mcgill.ca/?Topic=CasrSeqs2&v=new
Szalat A, Shpitzen S, Tsur A, Zalmon Koren I, Shilo S, Tripto-Shkolnik L et al (2017) Stepwise CaSR, AP2S1, and GNA11 sequencing in patients with suspected familial hypocalciuric hypercalcemia. Endocrine. 55(3):741–747
Eastell R, Brandi ML, Costa AG, D'Amour P, Shoback DM, Thakker RV (2014) Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab 99(10):3570–3579
Hannan FM, Thakker RV (2013) Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. 27(3):359–371
Tyler MR (2013) Control of renal calcium, phosphate, electrolyte, and water excretion by the calcium-sensing receptor. Best Pract Res Clin Endocrinol Metab 27(3):345–358
Hendy GN, D'Souza-Li L, Yang B, Canaff L, Cole DE (2000) Mutations of the calcium-sensing receptor (CASR) in familial hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat 16(4):281–296
Brown EM (2000) Familial hypocalciuric hypercalcemia and other disorders with resistance to extracellular calcium. Endocrinol Metab Clin N Am 29(3):503–522
Nissen PH, Christensen SE, Heickendorff L, Brixen K, Mosekilde L (2007) Molecular genetic analysis of the calcium sensing receptor gene in patients clinically suspected to have familial hypocalciuric hypercalcemia: phenotypic variation and mutation spectrum in a Danish population. J Clin Endocrinol Metab 92(11):4373–4379
CASR calcium sensing receptor [ Homo sapiens (human) ] Gene: NCBI; 2019 [updated July 30, 2019. Available from: https://www.ncbi.nlm.nih.gov/gene/846#top
Hu J, Reyes-Cruz G, Goldsmith PK, Spiegel AM (2001) The Venus's-flytrap and cysteine-rich domains of the human Ca2+ receptor are not linked by disulfide bonds. J Biol Chem 276(10):6901–6904
Hu J, Hauache O, Spiegel AM (2000) Human Ca2+ receptor cysteine-rich domain. Analysis of function of mutant and chimeric receptors. J Biol Chem 275(21):16382–16389
Lee S, Mannstadt M, Guo J, Kim SM, Yi HS, Khatri A et al (2015) A homozygous [Cys25]PTH(1-84) mutation that impairs PTH/PTHrP receptor activation defines a novel form of hypoparathyroidism. J Bone Miner Res 30(10):1803–1813
Hannan FM, Nesbit MA, Zhang C, Cranston T, Curley AJ, Harding B et al (2012) Identification of 70 calcium-sensing receptor mutations in hyper- and hypo-calcaemic patients: evidence for clustering of extracellular domain mutations at calcium-binding sites. Hum Mol Genet 21(12):2768–2778
Ward BK, Magno AL, Blitvich BJ, Rea AJ, Stuckey BG, Walsh JP et al (2006) Novel mutations in the calcium-sensing receptor gene associated with biochemical and functional differences in familial hypocalciuric hypercalcaemia. Clin Endocrinol 64(5):580–587
Marx SJ, Attie MF, Levine MA, Spiegel AM, Downs RW, Lasker RD (1981) The hypocalciuric or benign variant of familial hypercalcemia: clinical and biochemical features in fifteen kindreds. Medicine (Baltimore) 60(6):397–412
Aida K, Koishi S, Inoue M, Nakazato M, Tawata M, Onaya T (1995) Familial hypocalciuric hypercalcemia associated with mutation in the human Ca(2+)-sensing receptor gene. J Clin Endocrinol Metab 80(9):2594–2598
Taylor EN, Curhan GC (2009) Demographic, dietary, and urinary factors and 24-h urinary calcium excretion. Clinical journal of the American Society of Nephrology : CJASN 4(12):1980–1987
Pak CY, Sakhaee K, Moe OW, Poindexter J, Adams-Huet B, Pearle MS et al (2011) Defining hypercalciuria in nephrolithiasis. Kidney Int 80(7):777–782
Sakhaee K, Baker S, Zerwekh J, Poindexter J, Garcia-Hernandez PA, Pak CY (1994) Limited risk of kidney stone formation during long-term calcium citrate supplementation in nonstone forming subjects. J Urol 152(2 Pt 1):324–327
Christensen SE, Nissen PH, Vestergaard P, Heickendorff L, Brixen K, Mosekilde L (2008) Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clin Endocrinol 69(5):713–720
Jones AN, Shafer MM, Keuler NS, Crone EM, Hansen KE (2012) Fasting and postprandial spot urine calcium-to-creatinine ratios do not detect hypercalciuria. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 23(2):553–562
Cailleux A, Vuillermet P, Basuyau JP, Ménard JF, Lefebvre H, Kuhn JM, et al. A step towards cinacalcet testing for the diagnosis of primary hyperparathyroidism: comparison with the standardized intravenous calcium loading. A pilot study. Clin Endocrinol 2015;82(5):663-669
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
University of Calgary ethical approval was given for publication of biochemical data for a local control population.
The article does not contain any studies with animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants (patient and her son) included in the study.
Conflicts of interest
None.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 77 kb)
Rights and permissions
About this article

Cite this article
Mahajan, A., Buse, J. & Kline, G. Parathyroid hormone-dependent familial hypercalcemia with low measured PTH levels and a presumptive novel pathogenic mutation in CaSR. Osteoporos Int 31, 203–207 (2020). https://doi.org/10.1007/s00198-019-05170-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-019-05170-9