
Abstract
Methionine oxidation and reduction is a common phenomenon occurring in biological systems under both physiological and oxidative-stress conditions. The levels of methionine sulfoxide (MetO) are dependent on the redox status in the cell or organ, and they are usually elevated under oxidative-stress conditions, aging, inflammation, and oxidative-stress related diseases. MetO modification of proteins may alter their function or cause the accumulation of toxic proteins in the cell/organ. Accordingly, the regulation of the level of MetO is mediated through the ubiquitous and evolutionary conserved methionine sulfoxide reductase (Msr) system and its associated redox molecules. Recent published research has provided new evidence for the involvement of free MetO or protein-bound MetO of specific proteins in several signal transduction pathways that are important for cellular function. In the current review, we will focus on the role of MetO in specific signal transduction pathways of various organisms, with relation to their physiological contexts, and discuss the contribution of the Msr system to the regulation of the observed MetO effect.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Exposure of proteins to reactive oxygen species (ROS) and hydrogen peroxide under physiological and pathological conditions may lead to the oxidation of free methionine and methionyl residue, forming methionine sulfoxide (MetO) (Brot et al. 1981; Moskovitz et al. 1996). The function and structure of proteins that undergo this posttranslational modification may be altered, causing them to affect cellular function. The methionine sulfoxide reductase (Msr) system can reduce MetO to methionine, lowering the levels of MetO moiety of MetO-containing proteins resulting in changes to the function of these proteins (Oien and Moskovitz 2008). The Msr system consists of two families of enzymes: MsrA and MsrB, which are stereospecific and reduce S-MetO and R-MetO, respectively (Moskovitz et al. 2000; Grimaud 2001; Lowther et al. 2002; Moskovitz et al. 2002; Bar-Noy and Moskovitz 2002). Most mammals and yeast exhibit one type of MsrA and three types of MsrB (MsrB1-3) enzymes, while the types of Msr enzymes vary in bacteria and plants (Oien and Moskovitz 2008; Jiang and Moskovitz 2018). These enzymes require redox agents for their reducing activity, such as the thioredoxin/thioredoxin reductase systems, and the relative expression level of each type determines the total percent reduction of MetO (i.e., the sum of both MetO forms). The roles of both MetO and Msr system in health and disease have been investigated in various organisms from bacteria to mammals, including humans. For example, in mammals, a compromised Msr system and elevated levels of MetO are involved in the expression of markers that are associated with neurodegenerative diseases (i.e. Alzheimer’s and Parkinson’s diseases) (Jiang and Moskovitz 2018; Oien and Moskovitz 2019; Bitan et al. 2003; Dong et al. 2003; Boutte et al. 2006; Butterfield et al. 2005; Triguero et al. 2008; Wassef et al. 2007; Liu et al. 2008; Glaser et al. 2005), liver and kidney toxicity (Singh et al. 2017a, b; Noh et al. 2017), cancer (He et al. 2018; Kwak et al. 2017; Morel et al. 2017a, b), hearing loss (Ahmed et al. 2011; Kwon et al. 2014), mental health disorders (Campos et al. 2020; Walss-Bass et al. 2009; Ma et al. 2011; Otte et al. 2014), cardiovascular disease (García-Bermúdez et al. 2012; Rose et al. 2015; Gu et al. 2013), cystic fibrosis (Chandler et al. 2018; Magon et al. 2015; Knowles et al. 2012), and macular degeneration (Sreekumar et al. 2011; Brennan et al. 2009; Sreekumar et al. 2005). The role of free MetO in signaling pathways is not clear yet. The main reasons for this deficiency in knowledge are that free MetO is not associated with a specific protein that can be monitored for its function; and that the MetO level depends on the overall cellular redox state that it is not regulated only by the function of the Msr system. The burgeoning evidence supports the view that an enhanced function of the Msr system, which results in lower MetO levels, increases the cellular resistance to oxidative stress and related diseases (Jiang and Moskovitz 2018; Oien and Moskovitz 2019). However, the knowledge is limited regarding cell signaling that is mediated through the MetO moiety and the signal transduction pathways that affect both the Msr system and downstream events. Accordingly, in this review, we will describe and discuss the inter- and intra-relationships between MetO, Msr system, and recently associated cellular pathways. The function of the Msr system and formation of the MetO moiety are both processes that are ubiquitous in nature, while the current information on their association with signal transduction pathways per one species/organism is limited. Thus, the presented topics are sorted under categories according to the type of signaling, rather than the type of the biological system that is being investigated (i.e. type of organism).
Methionine sulfoxide-related signaling
Methionine sulfoxide and phosphorylation
Oxidation of methionine residues in proteins may change the protein structure–function properties, as it has been reported in many studies (Oien and Moskovitz 2008; Jiang and Moskovitz 2018; Oien and Moskovitz 2019). Likewise, phosphorylation and dephosphorylation of a protein may serve as protein modifiers that can alter its function and lead to a cascade of downstream events associated with signal transduction pathways. Thus, an obvious question arose: is there a connection and crosstalk between methionine oxidation and phosphorylation events of proteins? Several research studies aimed in answering this question by investigating how MetO residues can affect the phosphorylation rate of a specific protein. Hardin and colleagues showed that oxidation of methionine residue that is located within a hydrophobic region of a protein can inhibit protein phosphorylation in vitro (Hardin et al. 2009). Accordingly, this study demonstrated that the in vitro phosphorylation of a recombinant soybean calcium-dependent protein kinase and human AMP-dependent protein kinase was inhibited upon the oxidation of methionine residues. Further investigations revealed that methionine oxidation might inhibit protein phosphorylation in vivo as well, as it has been demonstrated for the Arabidopsis leaf nitrate reductase protein on Ser534.
These data support the suggestion that oxidation of enzyme-accessible MetO residues of proteins can inhibit the phosphorylation of adjacent phosphorylation sites, linking oxidative signals to alterations in protein phosphorylation through methionine oxidation. Another example for the negative effect of methionine oxidation on protein phosphorylation is the observation by Oien and researchers of the inhibitory effect of methionine oxidation on α-synuclein phosphorylation as it was observed in in vitro and ex vivo systems (Oien et al. 2009, 2011). Additional examples providing supportive evidence for the possible interaction between MetO residues and phosphorylation events are the correlation between Met293 oxidation and Ser292 phosphorylation of pyruvate dehydrogenase (Miernyk et al. 2009) and the correlation between Met45 oxidation and Ser32/36 phosphorylation of inhibitor kappa B alpha (Kanayama et al. 2002). In both examples, oxidation of the methionine residue inhibits phosphorylation of these serine residues.
Methionine sulfoxide as a signaling molecule in the methionine sulfoxide reductase system
The Msr system is able to reduce free and protein-bound MetO and plays an important role in cellular protein stability, function, and regulation (Oien and Moskovitz, 2008). Thus, during a state of excessive oxidative stress in cells, activation of the Msr system is important to lower the levels of MetO by reduction of MetO to methionine. This objective can be achieved both by an upregulation or activation of the Msr enzymes. One possible way to upregulate Msr is through molecules or peptides that exhibit a MetO moiety. For example, elevated MsrA activity and mRNA levels were observed in human neuroblastoma (IMR-32) cells in response to treatment with MetO-beta-amyloid (1–42) (Aβ42-MetO), suggesting that the cells exhibit a sensory system that can detect the presence of MetO in Aβ that, in turn, upregulates the expression of MsrA (Misiti et al. 2010). This observation has been also validated in mouse and rat neuronal cells that were grown in culture in the presence of Aβ42-MetO, Aβ40-MetO, or acetylated MetO molecules (Moskovitz et al. 2011). Other molecules that contain either a methyl group that can be oxidized or a methionine derivative moiety may also serve as Msr expression inducers. For example, the drug pergolide/pergolide sulfoxide contains a methyl group that when oxidized, either in vitro or cellular oxidation, can upregulate Msr activity and expression in cultured neuronal cells (Franklin et al. 2013). It was suggested that since pergolide and pergolide sulfoxide are dopamine receptor agonists and ligands for serotonin receptor their functions could be mediated through binding to these receptors, leading to the activation of a signal transduction pathway that regulates Msr expression. Additionally, it was discovered that S-adenosyl-methionine could cause a similar effect (Franklin et al. 2013). These compounds can cross the blood–brain barrier, and thus, it was proposed that they might be useful in the treatment of neurodegenerative diseases in which upregulation of the Msr system could be beneficiary due to its antioxidant capacity.
Effect of the Msr system on signal transduction
Calcium homeostasis and calcium-binding proteins
Calcium plays an important and significant role in many signal transduction pathways. The cellular level of calcium is strongly linked to the interaction between calcium and calmodulin. The calcium/calmodulin (Ca2+/CaM)-dependent protein kinase II (CaMKII) facilitates an increase of Ca2+ to cellular responses in excitable cells, and its activation is mediated through the Ca2+/CaM complex under physiological conditions (Erickson et al. 2008). However, both in vitro and in vivo oxidation of methionine residues of CaMKII cause the enzyme to remain constantly active, affecting several downstream cellular pathways (Erickson et al. 2008). In turn, this situation may lead to a compromised cardiac function if the MetO residues of Ca2+/CaM are not reduced to methionine by MsrA (Erickson et al. 2008). The CaM protein itself is prone to methionine oxidation that alters its function. Upon oxidation of specific methionine residues of CaM, the protein binds tightly to its target protein causing a lasting inhibitory effect that leads to down-regulation of energy metabolism in response to oxidative stress (Bigelow al. 2005). Similarly, oxidation of a methionine residue of the phospholamban protein (which regulates Ca-ATPase activity) is suggested to result in a tight binding of the protein to Ca-ATPase leading to a down-regulation of Ca-ATPase function in response to adrenergic signaling in the heart (Bigelow al. 2005). This inhibitory effect of methionine oxidation on energy metabolism can be salvaged by the Msr system, as it was demonstrated in Msr-overexpressed pancreatic stellate cells had an enhanced ATP-induced calcium response (Liu et al. 2019).
Methionine oxidation and reversal in inflammation
Oxidative stress in commonly accompanied by an inflammatory response in mammals, producing ROS and other tissue-damaging molecules. Thus, a strong antioxidant defense is supposed to protect and alleviate inflammatory-related insults to the organs/organism that are exposed to a high level of ROS. In that regard, upregulation and/or activation of the Msr system could play a positive role, both through the Msr enzymatic antioxidant activity and its ability to attenuate signal transduction pathways that are important for the inflammation process. Lipopolysaccharide (LPS) is known for its ability to induce a proinflammatory response. Indeed, silencing of MsrA expression in primary microglia cells caused an induction of microglia activation and the production of pro-inflammatory cytokines (Fan et al. 2015). Complementally, overexpression of recombinant MsrA in these microglia cells caused a reduction in the LPS-induced activation of p38 and ERK mitogen-activated protein kinases (MAPKs) and nuclear factor kappaB (NF-КB). Knocking out of the MsrA gene in mouse (MsrA KO) caused the animal to be hypersensitive to oxidative stress and exhibit phenotypes associated with neurodegeneration (Moskovitz et al. 2001). Recently, it was reported the MsrA KO mice possesses similar LPS-induced markers that were observed in the LPS-induced inflammation in glia cells (Fan et al. 2015). Taken together, it is concluded that MsrA plays an important role as a modulator of specific signal transduction pathways that are involved in the initiation of the inflammatory response, following LPS exposure. Interestingly, LPS specifically induces the expression of MsrB1 among all other Msrs (Singh et al. 2017a, b). Unexpectedly, ablation of MsrB1 caused a decreased induction of anti-inflammatory cytokines, such as interleukin (IL)-10 and the IL-1 receptor antagonist in the LPS-induced mice. This seemingly inconsistency with the proposed role of the Msr system in inflammation was adjusted by both the excessive pro-inflammatory cytokine production and an increase in acute tissue inflammation in the MsrB1 KO mice (Singh et al. 2017a, b). Apparently, the MsrB1 is also involved in the transcriptional regulation of dendritic cells upon LPS immunization. This is suggested by the observation that LPS-immunization induced MsrB1-dependent activation of the transcription-6 (STAT6) pathway and enhancement of IL-12 production, which promotes T-helper cells type 1 differentiation (Lee et al. 2017). Furthermore, MsrB1 promoted follicular helper T-cell differentiation, following immunization of the mice with sheep red blood cells (Lee et al. 2017).
Involvement of MsrA in mitochondrial function and glucose regulation
Oxidative stress is suggested to play an important role in maintaining glucose homeostasis. The observation that MsrA KO mice vulnerable to acquiring obesity-induced insulin resistance suggests that MsrA is involved in the regulation of related metabolic pathways. Accordingly, overexpression of recombinant MsrA in mitochondria indicated that the enzyme could alter glucose homeostasis following diet-induced obesity through the activating of AMPK signaling (Lee et al. 2020). This process ablated the insulin resistance caused by the diet although the obesity remained. Thus, it will be interesting and important to identify mitochondrial protein substrates for MsrA as means to discover how MsrA affects mitochondrial function in metabolic diseases.
Hyperglycemia can regulate angiogenesis through the induction of oxidative stress response and ROS. The RUNX2 DNA-binding transcription factor is activated by a glucose-mediated intracellular pathway, involved in endothelial cell function and angiogenesis, and is affected by oxidative stress. RUNX2 DNA-binding and endothelial cell differentiation are conserved in response to glucose and inhibited by hyperglycemia (mediated through elevated ROS production and the aldose reductase glucose-utilization pathway) (Hunnicut et al. 2015). The redox status of the methionine residues that regulates the RUNX2 DNA-binding has been found to be associated with the MsrA activity (Mochin et al. 2015). In addition, MsrA substrates and sulfoxide scavengers inhibited RUNX2 DNA binding in the absence of oxidative stress, while increasing this DNA binding in the presence of oxidants. Furthermore, MsrA was found to be associated with RUNX2:DNA complexes, and the homologue of RUNX2 protein, RUNX1, served also as a catalytic substrate for MsrA. The involvement of aldose reductase and MsrA in regulating RUNX2 transcription factor activity and the function of epithelial cells may lead to the development of novel therapies against vascular dysfunction that is associated with diabetes (Mochin et al. 2015).
Compromised mitochondrial respiration and cytochrome c oxidase activity (Complex IV) represent some of the phenotypes that are observed in an Alzheimer’s disease (AD) mouse model (Moskovitz et al. 2016). Ablation of the expression of MsrA in an AD-model mouse exacerbated these phenotypes, suggesting that the redox status of methionine residue/s of specific mitochondrial proteins may contribute to this phenomenon. Further investigations to identify mitochondrial MetO targets for MsrA may shed light into the role of MsrA in via MetO-dependent regulation of mitochondrial function (Moskovitz et al. 2016).
Role of the Msr system in protein degradation
Autophagy and mitophagy
Protein degradation is important for several processes that are required to maintain cellular metabolism and function. For example, the degradation system is involved in the following tasks: clearance of faulty proteins that cannot be salvaged to ensure proper cellular function; regulation of cell cycle through degradation of specific proteins that participate in controlling cell proliferation; and providing free amino acids energy production that is demanded under the extreme stressful conditions, such as starvation. Formation of MetO residue is a posttranslational modification that may lead to the cellular accumulation of MetO-proteins, if not reduced back to methionine by the Msr system. Thus, an open question remains: what is the role of Msr system in protein degradation that may prevent the occurrence of this phenomenon? Recent reports have provided some insight into the possible answers to this question. For example, deletion of the MsrA gene caused an increase in the production of p62-containing protein aggregates, activated autophagy, and decreased an apoptosis marker in vascular smooth muscle cells (Penningtona et al. 2018). Deletion of the MsrA gene in vascular smooth muscle cells enhances the interaction between Keap1 and p62 (Penningtona et al. 2018). One of the roles of Keap1 is to target the transcription factor, nuclear factor erythroid 2–related factor 2 (Nrf2) for proteasomal degradation (Nrf2 is important for the regulation of antioxidant genes). Thus, ablation of MsrA in vascular smooth muscle cells caused an inhibition of Nrf2 degradation through Keap1, since the availability of free Keap1 (i.e., not in a complex with p62) was diminished. This situation was reflected by the decreased ubiquitination of Nrf2 and an increased level of the Nrf2 protein. Consequently, the level of the Nrf2 in the nucleus was increased in the MsrA KO cells, leading to an upregulation of Nrf2-dependent transcriptional activity. In summary, these observations suggest a connection between autophagy and MsrA-dependent transcriptional activity and the expression level of Nrf2.
Mitophagy removes damaged mitochondria and protects the cell from apoptosis. ROS can damage mitochondria and the downstream events leading to mitophagy is yet to be fully understood. The role of the Msr system in oxidative stress-related mitophagy is not clear. Recently, it has been reported that the mitochondrial matrix protein MsrB2 participates in the process of initiating mitophagy by reducing the MetO moiety of the protein parkin (an E3-ubiquitine ligase) and prompting mitophagy through parkin-mediated ubiquitination and its interaction with LC3 (a key component of the autophagosomes) (Lee et al. 2019). This type of signaling depends on the presence of damaged mitochondria and the release of the MsrB2 protein from the mitochondria to the cytosol. Lack or inhibition of MsrB2 resulted in a reduced mitophagy and increased platelet apoptosis. Supportive evidence for the role of MsrB2 in promoting mitophagy is the observed correlation between an increased MsrB2 expression level in diabetes mellitus and increased level of platelet mitophagy, and reduced MsrB2 expression level and reduced mitophagy in platelets of Parkinson’s disease patients, respectively. Thus, MsrB2 is suggested to play an important role in the signaling process that facilitate the execution of oxidative stress-related mitophagy.
Ubiquitin and ubiquitin-like modifications
Ubiquitin (Ub) and ubiquitin-like (Ubl) modifications are part of the Ub/Ubl systems designed to target proteins for degradation by the ubiquitin–proteasome system. In a continuation of examining the role of the Msr system in autophagy and mitophagy (see the above section), the function of the Msr enzymes within the regulation of ubiquitination and Ubl processes is not clear. The first reported data that provided evidence for the involvement of MsrA in Ubl processes has been recently provided by Fu et al. (2017). These researchers observed that archaeal MsrA possess a Ubl protein modification activity in the presence of the Ubl-activating E1 (UbaA), in the presence of dimethyl sulfoxide (DMSO), and in the absence of reductant. Mass spectrometry analysis (LC–MS/MS) reveals that the formed MsrA-dependent Ubl conjugates are related to proteins that are associated with DNA replication, protein remodeling, and oxidative stress. These data provide a first glance at the specific role of MsrA (among all Msrs) in regulating Ubl modification in archaea under oxidative stress conditions.
To determine whether MsrA is involved in protein ubiquitination in mammals, the role of the mouse MsrA was investigated. Accordingly, it was discovered that the MsrA enzyme mediates the ubiquitination of the 14–3–3 zeta protein and promotes the binding of 14–3–3 proteins to alpha synuclein in brain (Deng et al. 2018). The 14–3–3 family of enzymes are involved in a variety of biological processes, including dopamine synthesis (through 14–3–3 zeta). The importance of MsrA in facilitating the ubiquitination of 14–3-3 zeta is manifested by the observation that lack of MsrA caused an upregulation of 14–3–3 zeta, leading to an enhanced dopamine level in the brain (Oien et al. 2008). Overall, it was concluded that MsrA-dependent 14–3–3 zeta ubiquitination affects the regulation of alpha synuclein degradation and dopamine synthesis in the brain (Deng et al. 2018).
MsrA plays an important role as a cellular antioxidant and promotes cell survival (Oien and Moskovitz 2019). The Ubl neddylation pathway, which is regulated by the c-Jun activation domain-binding protein-1 (Jab1), likewise affects cell survival (Zhou et al. 2019). Jab1 negatively regulates expression of the cell cycle inhibitor cyclin-dependent kinase inhibitor 1B (P27) by binding and targeting P27 for ubiquitination and degradation (Tomoda et al. 2002). Recently, we showed that MsrA interacts with Jab1 and enhances its deneddylase activity (removal of Nedd8) (Jiang et al. 2020). Consequently, the level of deneddylated Cullin-1 (Cul-1, a component of E3 Ub ligase complexes) was increased. Additionally, the action of MsrA increased the binding affinity of Jab1 to P27, while MsrA ablation caused a remarkable increase in the expression of P27 (Jiang et al. 2020). Thus, the positive regulation of MsrA on Jab1 function may serve to increase cellular resistance to oxidative stress and to promote cell survival.
Like MsrA, ablation of MsrB3 in mouse embryonic fibroblast (MEF) cells caused a decrease in cell proliferation (Lee et al. 2014). These MsrB3 KO cells also exhibited higher expression levels of the tumor protein 53 (p53), cyclin-dependent kinase inhibitor p21, and p27 in comparison to control cells. These data provide additional evidence for the involvement of Msr enzymes in cell-cycle regulation that is mediated by the Ub/Ubl -dependent degradation pathways.
Regulation of transcriptional factors based on the redox state of methionine
The integration of nitrate from a soil source is an important feature that is required for the survival of microorganisms and plants. Activation of the nitrate-specific transcription factor NirA in Aspergillus nidulans is a process that involves both nuclear retention of NirA and its conversion to a functional activator (Gallmetzer et al. 2015). Intracellular nitrate or nitrite leads to disruption of the interaction between the nuclear export sequence (NES) of NirA and the specific exportin KapK, the CRM1 homologue in A. nidulans. Consequently, NirA rapidly accumulates in the nucleus and is then able to bind to the UAS (upstream activating sequences) of genes involved in nitrate absorption. In the absence of nitrate, when NirA is inactive and, predominantly, present in the cytosol, Met169 of its nuclear export sequence (NES) is oxidized to MetO. This oxidation depends on the activity of the enzyme FmoB, a flavin-containing monooxygenase. However, exposure of A. nidulans cells to nitrate leads to a reduction of NirA-MetO to NirA-Met by a process that is independent of the Msr system. Accordingly, it was proposed that in the presence of nitrate, the activation domain is exposed and restricts the active NirA protein to the nucleus. However, in the absence of nitrate, Met169 is oxidized by an FmoB-dependent manner, causing a loss of NirA protection by its nitrate regulatory domain (NiRD), leading to the NES exposure, and consequently relocating of the inactive NirA to the cytosol. These complex events of transcription regulation are examples of the interaction between the accessibility of nitrate, methionine oxidation-dependent translocation of the NirA to the cytosol, and the transcriptional pathways leading to nitrate absorption.
Neuronal activation of c-Jun N-terminal kinase (JNK)/forkhead boxO(FOXO) transcription factors participate in the regulation of growth, metabolism, lifespan, and stress resistance in various organisms, including Drosophila (Chung et al. 2010). FOXO is regulated by the insulin signaling pathway and the stress-induced JNK signaling pathway. Oxidative stress activates JNK that translocates FOXO into the nucleus, causing an enhanced expression of antioxidant proteins. Among these antioxidants, the expression of the Drosophila’s MsrA was enhanced through the downstream signaling mediated by FOXO. As expected, the resulting expression of MsrA enhanced the Drosophila’s resistance to oxidative stress conditions and increased its survival rate. Furthermore, overexpression of MsrA in fat body cells caused FOXO to translocate to the nucleus. This latter observation suggests that MetO reversal to Met by Msr of proteins (including FOXO) promotes FOXO to translocate to the nucleus. This possible MetO-dependent regulation of FOXO may indicate a feedback relationship between MetO and MsrA and the FOXO-related transcriptional regulation of genes that are important for the survival of Drosophila.
In bacteria, Ffh (“fifty-four homologue”) protein contains a methionine‐rich region that interacts with a small 4.5S RNA. This interaction is not directly involved in transcriptional regulation but it is unique and can affect protein regulation. For example, oxidation of the methionine moiety of Ffh prevents it from binding to the 4.5S RNA and this function can be restored by the action of MsrA/B (Ezraty et al. 2004). In turn, lack of MsrA and MsrB leads to defect in the Ffh‐dependent targeting of Maltose/maltodextrin transport system permease protein (MalF).
The bacterial HypT (hypochlorite-responsive transcription factor) is activated by methionine oxidation to confer hypochlorite (HOCl) resistance. When activated, HypT regulates target genes and downregulates intracellular iron levels. This type of MetO-dependent transcriptional regulation provides bacterial cells the ability to survive under oxidative stress conditions (i.e. elevated HOCl levels) (Drazic et al. 2013).
Determinants that affect the expression levels of Msr enzymes
The physiological expression levels of the Msr enzymes are dependent on various factors. The MsrA expression level was shown to be regulated upon exposure of cells to several environmental conditions such as: an increase of the extracellular level of the MetO and methyl-containing compounds moiety in cultured mammalian cells (Misiti et al. 2010; Moskovitz et al. 2011; Franklin et al. 2013) stationary phase and starvation conditions of bacterial cells (Moskovitz et al. 1995), the presence of antibiotics in cultured bacterial cells (Singh et al. 2001), changes in UV radiation of human skin cells (Ogawa et al. 2006), exposure of human cell culture to the natural polyphenol resveratrol (Wu et al. 2013), and treatment of rats with copper (which also affected the expression of MsrB genes) (Zhong et al. 2021). The search for transcriptional regulation of MsrA by specific proteins revealed that there is a feedback relationship between MsrA and MsrB expression levels, as lack of one isotype downregulates its counterpart (Moskovitz and Stadtman 2003; Fomenko et al. 2009). More specifically, two proteins were identified in their ability to increase MsrA transcriptional expression: transcription factor FOXO in Caenorhabditis elegans and Drosophila melanogaster. (Minniti et al. 2009; Chung et al. 2010), and a homologue of elongation factor one-gamma (EF-1γ) with thioredoxin (Trx) in yeast (Hanbauer et al. 2003; Hanbauer and Moskovitz 2006).
The expression of the MsrB enzymes was also affected by environmental conditions, in which some of them share similarities with the conditions affecting MsrA expression. The information about MsrB transcription regulation in the scientific literature is limited, However, like MsrA, the expression of MsrB is also affected by antibiotics in bacterial cells (Baum et al. 2015) and the transcription level of MsrB1 (a selenoprotein) is reduced when mice are fed with a selenium-deficient diet (Moskovitz and Stadtman 2003). In plants, there are several subtypes of MsrA and MsrB gene family and their expression levels are changing in response to the environmental conditions such as oxidative and osmotic stress, mineral availability, light exposure, and drought (Rey et al. 2018).
Conclusion
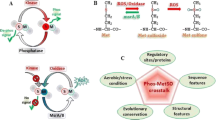
A summary figure of this review illustrates the involvement of MetO and the Msr enzymes in signal transduction pathways (Fig. 1). The cellular signaling that are prompted by either the action of free MetO, protein-bound MetO, or the Msr system, needs further investigation given the relatively limited knowledge available in this field. The described effects of MsrA/MsrB on cellular/organism signaling pathways are based on the demonstrated correlations between their expression and activity levels and the observed downstream cellular events. Accordingly, more research is needed to explore the complete fashion of these effects (i.e., direct or indirect consequences of MsrA/MsrB expression levels). Nonetheless, the expression and function of the MsrA/MsrB enzymes themselves seem to be more directly affected through selenium availability (for the selenoprotein MsrB1) and by their co-expression levels (MsrA and MsrB1 affect each other’s expression level). There are several research options that may expand our understanding of the processes that are involved in MetO/Msr system-related signal transduction pathways. For example, studies can focus on the exploration of specific epigenetics factors as potential signaling factors in the expression of phenomena associated with the MetO/Msr system. Among these factors, genomic methylation could play a role in oxidative-stress-related gene regulation that can be affected by the redox status of methionine. Because the major methyl donor in the cell is the amino acid methionine, it is predicted that excess level of free MetO may reduce the pool of free methionine and thus inhibit or change the selectivity of the gene methylation process. Furthermore, specific transcriptional factors are regulated by their cellular location (i.e., nucleus or cytosol). Therefore, the MetO levels or Ub/ Ubl modifications (regulated by the function or compromised function of the Msr system) of these factors may affect the cellular localization of these transcription factors and consequently lead to alterations in the progress of signal transduction pathways. Fostering extensive research in this field is predicted to provide the foundation for the development of new MetO/Msr-based therapies against pathologies that are associated with aging and oxidative-stress-related diseases.

A summary of the identified regulators affecting Msr enzymes and related substrate through signal transduction. The identification of positive and negative regulators on the expression of Msr enzymes (MsrA and MsrB types; Brot et al. 1981; Moskovitz et al. 1996; Oien and Moskovitz 2008) as follows. Positive regulators: Ultraviolet radiation (UV) (Ogawa et al. 2006), methionine sulfoxide (MetO) (Misiti et al. 2010; Moskovitz et al. 2011), the polyphenol resveratrol (Wu et al. 2013), copper (Zhong et al. 2021), the antibiotics Oxacillin (Singh et al. 2001), methyl-containing compounds (Franklin et al. 2013), transcription factor forkhead box O (FOXO) of Caenorhabditis elegans and Drosophila melanogaster (Minniti et al. 2009; Chung et al. 2010), a homologue of elongation factor one-gamma (EF-1γ) with thioredoxin (Trx) in yeast (Hanbauer et al. 2003; Hanbauer and Moskovitz 2006), and the counterpart enzyme for of each type of the Msr enzyme (i.e., MsrA or MsrB) (Moskovitz and Stadtman, 2003; Fomenko et al. 2009). It is important to note, that not all the factors that upregulate MsrA expression were tested for their ability to upregulate MsrB enzymes. Thus, their specificity towards MsrB should be examined. The only identified negative regulator for Msr expression is selenium that is needed both for the expression and function of MsrB1 (Bar-Noy and Moskovitz 2002; Moskovitz and Stadtman 2003). Following signal transduction events resulting from the reducing activity of either MsrA or MsrB on their substrates (known and yet to be discovered), changes in the function, cellular location, or expression of specific proteins or conditions were observed as indicated: Calcium regulation (Erickson et al. 2008; Bigelow et al. 2005; Liu et al. 2019); Inflammation (Fan et al. 2015; Singh et al. 2017a, b; Lee et al. 2017, 2020); Glucose homeostasis (Hunnicut et al. 2015; Mochin et al. 2015); Autophagy and mitophagy (Penningtona et al. 2018; Lee et al. 2019); Ubiquitin/Ubiquitin-like modifications (Ub/Ubl) in archaea and mouse (Fu et al. 207; Deng et al. 2018; Jiang et al. 2020); Transcriptional regulation in various organisms (Lee et al. 2014; Gallmetzer et al. 2015; Chung et al. 2010; Drazic et al. 2013; Chung et al. 2010; Hanbauer et al. 2003; Hanbauer and Moskovitz 2006). CaM calmodulin, CaMKII calmodulin kinase II, Nrf2 nuclear factor erythroid 2–related factor 2, Jab1 c-Jun activation domain-binding protein-1, P27 cyclin-dependent kinase inhibitors: P21, P27, P53 Tumor protein P53, LPS Lipopolysaccharide
References
Ahmed ZM, Yousaf R, Lee BC, Khan SN, Lee S, Lee K, Husnain T, Rehman AU, Bonneux S, Ansar M et al (2011) Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet 88:19–29
Bar-Noy S, Moskovitz J (2002) Mouse methionine sulfoxide reductase B: effect of selenocysteine incorporation on its activity and expression of the seleno-containing enzyme in bacterial and mammalian cells. Biochem Biophys Res Commun 297(4):956–961
Baum KR, Ahmad Z, Singh VK (2015) Regulation of expression of oxacillin-inducible methionine sulfoxide reductases in Staphylococcus aureus. Int J Microbiol 2015:617925
Bigelow DJ, Squier TC (2005) Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim Biophys Acta 1703:121–134
Bitan G, Tarus B, Vollers SS, Lashuel HA, Condron MM, Straub JE, Teplow DB (2003) A molecular switch in amyloid assembly: Met35 and amyloid beta-protein oligomerization. J Am Chem Soc 25:15359–15365
Boutte AM, Woltjer RL, Zimmerman LJ, Stamer SL, Montine KS, Manno MV, Cimino PJ, Liebler DC, Montine TJ (2006) Selectively increased oxidative modifications mapped to detergent-insoluble forms of Abeta and beta-III tubulin in Alzheimer’s disease. FASEB J 20:1473–1483
Brennan LA, Kantorow M (2009) Mitochondrial function and redox control in the aging eye: role of MsrA and other repair systems in cataract and macular degenerations. Exp Eye Res 88(2):195–203
Brot N, Weissbach L, Werth J, Weissbach H (1981) Enzymatic reduction of protein-bound methionine sulfoxide. Proc Natl Acad Sci U S A 78(4):2155–2158
Butterfield DA, Boyd-Kimball D (2005) The critical role of methionine 35 in Alzheimer’s amyloid beta-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta 1703:149–156
Campos AI, García-Marín LM, Byrne EM, Martin NG, Cuéllar-Partida G, Rentería ME (2020) Insights into the aetiology of snoring from observational and genetic investigations in the UK Biobank. Nat Commun 11(1):817–829
Chandler JD, Margaroli C, Horati H, Kilgore MB, Veltman M, Liu HK, Taurone AJ, Peng L, Guglani L, Uppal K, Go YM, Tiddens HAWM, Scholte BJ, Tirouvanziam R, Jones DP, Janssens HM (2018) Myeloperoxidase oxidation of methionine associates with early cystic fibrosis lung disease. Eur Respir J 52(4):1801118
Chung H, Kim AK, Jung SA, Kim SW, Yu K, Lee JH (2010) The Drosophila homolog of methionine sulfoxide reductase A extends lifespan and increases nuclear localization of FOXO. FEBS Lett 584:3609–3614
Deng Y, Jiang B, Rankin CL, Toyo-Oka K, Richter ML, Maupin-Furlow JA, Moskovitz J (2018) Methionine sulfoxide reductase A (MsrA) mediates the ubiquitination of 14–3–3 protein isotypes in brain. Free Radic Biol Med 129:600–607
Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR (2003) Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42:2768–2773
Drazic A, Miura H, Peschek J, Le Y, Bach NC, Kriehuber T, Winter J (2013) Methionine oxidation activates a transcription factor in response to oxidative stress. Proc Natl Acad Sci U S A 110(23):9493–9498
Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE, Aykin-Burns N et al (2008) A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell 133:462–474
Ezraty B, Grimaud R, El Hassouni M, Moinier D, Barras F (2004) Methionine sulfoxide reductases protect Ffh from oxidative damages in Escherichia coli. EMBO J 23(8):1868–1877
Fan H, Wu PF, Zhang L, Hu ZL, Wang W, Guan XL, Luo H, Ni M, Yang JW, Li MX, Chen JG, Wang F (2015) Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammation via inhibiting ROS/MAPKs/NF-κB signaling pathways through a catalytic antioxidant function. Antioxid Redox Signal 22(10):832–847
Fomenko DE, Novoselov SV, Natarajan SK, Lee BC, Koc A, Carlson BA, Lee TH, Kim HY, Hatfield DL, Gladyshev VN (2009) MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J Biol Chem 284(9):5986–5993
Franklin JM, Carrasco GA, Moskovitz J (2013) Induction of methionine sulfoxide reductase activity by pergolide, pergolide sulfoxide, and S-adenosyl-methionine in neuronal cells. Neurosci Lett 533:86–89
Fu X, Adams Z, Liu R, Hepowit NL, Wu Y, Bowmann CF, Moskovitz J, Maupin-Furlow JA (2017) Methionine sulfoxide reductase A (MsrA) and its function in ubiquitin-like protein modification in archaea. Mbio 8(5):e01169-e1217. https://doi.org/10.1128/mBio.01169-17
Gallmetzer A, Silvestrini L, Schinko TB, Hortschansky P, Dattenböck C, Muro-Pastor MI, Kungl A, Brakhage AA, Scazzocchio C, Strauss J (2015) Reversible oxidation of a conserved methionine in the nuclear export sequence determines subcellular distribution and activity of the fungal nitrate regulator NirA. PLoS Genet 11(7):e1005297
García-Bermúdez M, López-Mejías R, González-Juanatey C, Castañeda S, Miranda-Filloy JA, Blanco R, Fernández-Gutiérrez B, Balsa A, González-Álvaro I, Gómez-Vaquero C, Llorca J, Martín J, González-Gay MA (2012) Association of the methionine sulfoxide reductase A rs10903323 gene polymorphism with cardiovascular disease in patients with rheumatoid arthritis. Scand J Rheumatol 41(5):350–353
Glaser CB, Yamin G, Uversky VN, Fink AL (2005) Methionine oxidation, alpha-synuclein and Parkinson’s disease. Biochim Biophys Acta 1703(2):157–169
Grimaud R, Ezraty B, Mitchell JK, Lafitte D, Briand C, Derrick PJ, Barras F (2001) Repair of oxidized proteins. Identification of a new methionine sulfoxide reductase. J Biol Chem 276(52):48915–48920
Gu H, Chen W, Yin J, Chen S, Zhang J, Gong J (2013) Methionine sulfoxide reductase A rs10903323 G/A polymorphism is associated with increased risk of coronary artery disease in a Chinese population. Clin Biochem 46(16–17):1668–1672
Hanbauer I, Moskovitz J (2006) The yeast cytosolic thioredoxins are involved in the regulation of methionine sulfoxide reductase A. Free Radic Biol Med 40(8):1391–1396
Hanbauer I, Boja ES, Moskovitz J (2003) A homologue of elongation factor 1 gamma regulates methionine sulfoxide reductase A gene expression in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 100(14):8199–8204
Hardin SC, Larue CT, Oh MH, Jain V, Huber SC (2009) Coupling oxidative signals to protein phosphorylation via methionine oxidation in Arabidopsis. Biochem J 422(2):305–312
He Q, Li H, Meng F, Sun X, Feng X, Chen J, Li L, Liu J (2018) Methionine sulfoxide reductase B1 regulates hepatocellular carcinoma cell proliferation and invasion via the mitogen-activated protein kinase pathway and epithelial-mesenchymal transition. Oxid Med Cell Longev 2018:5287971
Hunnicut JL, Liu Y, Richardson A, Salmon AB (2015) MsrA overexpression targeted to the mitochondria, but not cytosol, preserves insulin sensitivity in diet-induced obese mice. PLoS ONE 10(10):e0139844
Jiang B, Moskovitz J (2018) The functions of the mammalian methionine sulfoxide reductase system and related diseases. Antioxidants (basel) 7(9):E122. https://doi.org/10.3390/antiox7090122
Jiang J, Adams Z, Moonah S, Shi S, Furlow JM, Moskovitz J (2020) The antioxidant enzyme methionine sulfoxide reductase A (MsrA) interacts with Jab1/CSN5 and regulates its function. Antioxidants (basel) 9(5):452–469
Kanayama A, Inoue J, Sugita-Konishi Y, Shimizu M, Miyamoto Y (2002) Oxidation of IκBα at methionine 45 is one cause of taurine chloramine-induced inhibition of NF-κB activation. J Biol Chem 277:24049–24056
Knowles MR, Drumm M (2012) The influence of genetics on cystic fibrosis phenotypes. Cold Spring Harb Perspect Med 2(12):a009548
Kwak GH, Kim HY (2017) MSRB3 deficiency induces cancer cell apoptosis through p53-independent and ER stress-dependent pathways. Arch Biochem Biophys 621:1–5
Kwon TJ, Cho HJ, Kim UK, Lee E, Oh SK, Bok J, Bae YC, Yi JK, Lee JW, Ryoo ZY et al (2014) Methionine sulfoxide reductase B3 deficiency causes hearing loss due to stereocilia degeneration and apoptotic cell death in cochlear hair cells. Hum Mol Genet 23:1591–1601
Lee E, Kwak GH, Kamble K, Kim HY (2014) Methionine sulfoxide reductase B3 deficiency inhibits cell growth through the activation of p53–p21 and p27 pathways. Arch Biochem Biophys 547:1–5
Lee BC, Lee SG, Choo MK, Kim JH, Lee HM, Kim S, Fomenko DE, Kim HY, Park JM, Gladyshev VN (2017) Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci Rep 7(1):5119–5128
Lee SH, Lee S, Du J, Jain K, Ding M, Kadado AJ, Atteya G, Jaji Z, Tyagi T, Kim WH, Herzog RI, Patel A, Ionescu CN, Martin KA, Hwa J (2019) Mitochondrial MsrB2 serves as a switch and transducer for mitophagy. EMBO Mol Med 11(8):e10409
Lee HJ, Park JS, Yoo HJ, Lee HM, Lee BC, Kim JH (2020) The selenoprotein MsrB1 instructs dendritic cells to induce T-helper 1 immune responses. Antioxidants (basel) 9(10):1021–1039
Liu JS, Cui ZJ (2019) Pancreatic stellate cells serve as a brake mechanism on pancreatic acinar cell calcium signaling modulated by methionine sulfoxide reductase expression. Cells 8(2):109–133
Liu F, Hindupur J, Nguyen JL, Ruf KJ, Zhu J, Schieler JL, Bonham CC, Wood KV, Davisson VJ, Rochet JC (2008) Methionine sulfoxide reductase A protects dopaminergic cells from Parkinson’s disease-related insults. Free Radic Biol Med 45:242–255
Lowther WT, Weissbach H, Etienne F, Brot N, Matthews BW (2002) The mirrored methionine sulfoxide reductases of Neisseria gonorrhoeae pilB. Nat Struct Biol 9(5):348–352
Ma X, Deng W, Liu X, Li M, Chen Z, He Z, Wang Y, Wang Q, Hu X, Collier DA, Li T (2011) A genome-wide association study for quantitative traits in schizophrenia in China. Genes Brain Behav 10(7):734–739
Magon NJ, Turner R, Gearry RB, Hampton MB, Sly PD, Kettle AJ (2015) Oxidation of calprotectin by hypochlorous acid prevents chelation of essential metal ions and allows bacterial growth: relevance to infections in cystic fibrosis. Free Radic Biol Med 86:133–144
Miernyk JA, Johnston ML, Huber SC, Tovar-Méndez A, Hoyos E, Randal DD (2009) Oxidation of an adjacent methionine residueinhibits regulatory seryl-phosphorylation of pyruvate dehydrogenase. Proteom Insights 2:15–22
Minniti AN, Cataldo R, Trigo C, Vasquez L, Mujica P, Leighton F, Inestrosa NC, Aldunate R (2009) Methionine sulfoxide reductase A expression is regulated by the DAF-16/FOXO pathway in Caenorhabditis elegans. Aging Cell 8(6):690–705
Misiti F, Clementi ME, Giardina B (2010) Oxidation of methionine 35 reduces toxicity of the amyloid β-peptide(1–42) in neuroblastoma cells (IMR-32) via enzyme methionine sulfoxide reductase A expression and function. Neurochem Int 56:597–602
Mochin MT, Underwood KF, Cooper B, McLenithan JC, Pierce AD, Nalvarte C, Arbiser J, Karlsson AI, Moise AR, Moskovitz J, Passaniti A (2015) Hyperglycemia and redox status regulate RUNX2 DNA-binding and an angiogenic phenotype in endothelial cells. Microvasc Res 7:55–64
Morel AP, Ginestier C, Pommier RM, Cabaud O, Ruiz E, Wicinski J, Devouassoux-Shisheboran M, Combaret V, Finetti P, Chassot C et al (2017a) A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome. Nat Med 23:568–578
Morel AP, Ginestier C, Pommier RM, Cabaud O, Ruiz E, Wicinski J, Devouassoux-Shisheboran M, Combaret V, Finetti P, Chassot C et al (2017b) A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome. Nat Med 23:568–578
Moskovitz J, Stadtman ER (2003) Selenium-deficient diet enhances protein oxidation and affects methionine sulfoxide reductase (MsrB) protein level in certain mouse tissues. Proc Natl Acad Sci USA 100(13):7486–7490
Moskovitz J, Rahman MA, Strassman J, Yancey SO, Kushner SR, Brot N, Weissbach H (1995) Escherichia coli peptide methionine sulfoxide reductase gene: regulation of expression and role in protecting against oxidative damage. J Bacteriol 177(3):502–507
Moskovitz J, Weissbach H, Brot N (1996) Cloning the expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proc Natl Acad Sci U S A 93(5):2095–2099
Moskovitz J, Poston M, Berlett BS, Nosworthy JN, Szczepanowski R, Stadtman ER (2000) Identification and characterization of a putative active site for peptide-methionine sulfoxide reductase (MsrA) and its substrate stereospecificity. J Biol Chem 275:14167–14172
Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER (2001) Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci U S A 98(23):12920–12925
Moskovitz J, Singh VK, Requena J, Wilkinson BJ, Jayaswal RK, Stadtman ER (2002) Purification and characterization of methionine sulfoxide reductases from mouse and Staphylococcus aureus and their substrate stereospecificity. Biochem Biophys Res Commun 290(1):62–65
Moskovitz J, Maiti P, Lopes DH, Oien DB, Attar A, Liu T, Mittal S, Hayes J, Bitan G (2011) Induction of methionine-sulfoxide reductases protects neurons from amyloid β-protein insults in vitro and in vivo. Biochemistry 50(49):10687–10697
Moskovitz J, Du F, Bowman CF, Yan SS (2016) Methionine sulfoxide reductase A affects -amyloid solubility and mitochondrial function in a mouse model of Alzheimer’s disease. Am J Physiol Endocrinol Metab 310:E388–E393
Noh MR, Kim KY, Han SJ, Kim JI, Kim HY, Park KM (2017) Methionine sulfoxide Reductase A deficiency exacerbates cisplatin-induced nephrotoxicity via increased mitochondrial damage and renal cell death. Antioxid Redox Signal 27:727–741
Ogawa F, Sander CS, Hansel A, Oehrl W, Kasperczyk H, Elsner P, Shimizu K, Heinemann SH, Thiele LL (2006) The repair enzyme peptide methionine-S-sulfoxide reductase is expressed in human epidermis and upregulated by UVA radiation. J Invest Dermatol 126(5):1128–1134
Oien DB, Moskovitz J (2008) Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr Top Dev Biol 80:93–133
Oien DB, Moskovitz J (2019) Genetic regulation of longevity and age-associated diseases through the methionine sulfoxide reductase system. Biochim Biophys Acta Mol Basis Dis 1865(7):1756–1762
Oien DB, Osterhaus GL, Latif SA, Pinkston JW, Fulks J, Johnson MA, Fowler SC, Moskovitz J (2008) MsrA knockout mouse exhibits abnormal behavior and brain dopamine levels. Free Radic Biol Med 45(2):193–200
Oien DB, Shinogle HE, Moore DS, Moskovitz J (2009) Clearance and phosphorylation of alpha-synuclein are inhibited in methionine Sulfoxide reductase A null Yeast cells. J Mol Neuroscience 39(3):323–332
Oien DB, Carrasco GA, Moskovitz J (2011) Decreased phophorylation and increased methionine oxidation of α-synuclein in the methionine sulfoxide reductase A knockout mouse. J Amino Acids (Article ID, 721094)
Otte DM, Raskó T, Wang M, Dreiseidler M, Drews E, Schrage H, Wojtalla A, Höhfeld J, Wanker E, Zimmer A (2014) Identification of the mitochondrial MSRB2 as a binding partner of LG72. Cell Mol Neurobiol 34(8):1123–1130
Penningtona SM, Kluthoa PR, Xiea L, Broadhursta K, Kovala OM, McCormickb ML, Spitzb DR, Grumbacha IM (2018) Defective protein repair under methionine sulfoxide A deletion drives autophagy and ARE-dependent gene transcription. Red Biol 16:401–408
Rey P, Tarrago I (2018) Physiological roles of plant methionine sulfoxide reductases in redox homeostasis and signaling. Antioxidants (basel) 7(9):114. https://doi.org/10.3390/antiox7090114
Rose AH, Hoffmann PR (2015) Selenoproteins and cardiovascular stress. Thromb Haemost 113(3):494–504
Singh VK, Moskovitz J, Wilkinson BJ, Jayaswal RK (2001) Staphylococcus aureus that contributes to oxidative defence and is highly induced by the cell-wall-active antibiotic oxacillin. Microbiology 147(Pt 11):3037–3045
Singh MP, Kwak GH, Kim KY, Kim HY (2017a) Methionine sulfoxide reductase A protects hepatocytes against acetaminophen-induced toxicity via regulation of thioredoxin reductase 1 expression. Biochem Biophys ResCommun 487:695–701
Singh MP, Kim KY, Kwak GH, Baek SH, Kim HY (2017b) Methionine sulfoxide reductase A protects against lipopolysaccharide-induced septic shock via negative regulation of the proinflammatory responses. Arch Biochem Biophys 631:42–48
Sreekumar PG, Kannan R, Yaung J, Spee CK, Ryan SJ, Hinton DR (2005) Protection from oxidative stress by methionine sulfoxide reductases in RPE cells. Biochem Biophys Res Commun 334(1):245–253
Sreekumar PG, Hinton DR, Kannan R (2011) Methionine sulfoxide reductase A: Structure, function and role in ocular pathology. World J Biol Chem 2(8):184–192
Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY (2002) The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem 277:2302–2310
Triguero L, Singh R, Prabhakar R (2008) Comparative molecular dynamics studies of wild-type and oxidized forms of full-length Alzheimer amyloid beta-peptides Abeta (1–40) and Abeta (1–42). J Phys Chem B 112:7123–7131
Walss-Bass C, Soto-Bernardini MC, Johnson-Pais T, Leach RJ, Ontiveros A, Nicolini H, Mendoza R, Jerez A, Dassori A, Chavarria-Siles I, Escamilla MA, Raventos H (2009) Methionine sulfoxide reductase: a novel schizophrenia candidate gene. Am J Med Genet B Neuropsychiatr Genet 150B(2):219–225
Wassef R, Haenold R, Hansel A, Brot N, Heinemann SH, Hoshi T (2007) Methionine sulfoxide reductase A and a dietary supplement S-methyl-l-cysteine prevent Parkinson’s-like symptoms. J Neurosci 27:12808–12816
Wu PF, Xie N, Zhang JJ, Guan XL, Zhou J, Long LH, Li YL, Xiong QJ, Zeng JH, Wang F, Chen JG (2013) Resveratrol preconditioning increases methionine sulfoxide reductases A expression and enhances resistance of human neuroblastoma cells to neurotoxins. J Nutr Biochem 24(6):1070–1077
Zhong G, He Y, Wan F, Wu S, Jiang X, Tang Z, Hu L (2021) Effects of long-term exposure to copper on the Keap1/Nrf2 signaling pathway and Msr-related redox status in the kidneys of rats. Biol Trace Elem Res. https://doi.org/10.1007/s12011-020-02557-2
Zhou L, Jiang Y, Luo Q, Li L, Jia L (2019) Neddylation: a novel modulator of the tumor microenvironment. Mol Cancer 18:77–88
Funding
This research received no external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethics approval (include appropriate approvals or waivers)
Not applicable.
Consent to participate (include appropriate statements)
Not applicable.
Consent for publication (include appropriate statements)
Not applicable.
Availability of data and material (data transparency)
Not applicable.
Code availability (software application or custom code)
Not applicable.
Additional information
Handling editor: H. Jakubowski.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Moskovitz, J., Smith, A. Methionine sulfoxide and the methionine sulfoxide reductase system as modulators of signal transduction pathways: a review. Amino Acids 53, 1011–1020 (2021). https://doi.org/10.1007/s00726-021-03020-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-021-03020-9