
Abstract
l-Homoarginine is a cationic amino acid derivative, which is structurally related to l-arginine and lysine. Several lines of evidence point to nervous tissue as an important target of homoarginine action. In the mammalian brain homoarginine can be detected in noticeable quantities, but its origin is currently poorly explored. In part I of this review we try to show that both uptake and transport into brain (carried out by cationic amino acid transporters) and local synthesis in the brain (carried out by the homoarginine-synthesizing enzymes l-arginine:glycine amidinotransferase and ornithine transcarbamylse) might contribute to homoarginine brain content. We then give a brief overview about the multiple effects of homoarginine on the healthy brain and show that both homoarginine excess and deficiency are potentially harmful to the central nervous system. In part II, we shortly report about own experiments with regard to the cellular localization of cationic amino acid transporters, as well the enzymes l-arginine:glycine amidinotransferase and ornithine transcarbamylse, in human and rat brains.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Part I: Review of the literature
Homoarginine: brief outline of its career
l-Homoarginine (hArg) is a cationic, non-proteinogenic amino acid derivative, which is structurally related to l-arginine and lysine. It is derived from lysine by enzymatic guanidination of lysine residues.
Historically, it has to be stated that after its successful artificial synthesis by Steib (1926) and the subsequent identification as a naturally occurring substance (Stevens and Bush 1950), hArg for a number of years lived a wallflower existence with regard to research activities. This situation changed when it became evident that hArg (1) is synthesized in, and released from, liver and kidney tissues (Ryan et al. 1968, 1969), (2) circulates in the blood stream and (3) acts as an effective inhibitor of alkaline phosphatase isoenzymes in serum, liver, intestine, amniotic fluid, placenta and bone (Fishman and Sie 1971 and many others), thereby being able, for example, to delay the growth of osteosarcoma tumor cells (Kikuchi et al. 1982). Interest in possible physiological and pathophysiological roles of hArg further increased after an influential paper by Lambert et al. (1992), demonstrating that hArg can serve as a substrate for nitric oxide (NO) synthesis by brain, macrophage and endothelium nitric oxide synthases (i.e., NOS isoforms nNOS, iNOS and eNOS). Two of the most striking recent discoveries were on hArg’s implication for bone metabolism and for normal and pathologic cardiac function. It was shown that abnormally low hArg concentrations not only accelerate bone turnover (Pilz et al. 2012), but are also a risk factor for heart disease (Pilz et al. 2011), with hArg emerging as a suitable biomarker for the outcome of cardiovascular diseases (reviewed by Atzler et al. 2015). Over time much about hArg’s functions has been learned from patients with hyperargininemia. Hyperargininemia is a rare autosomal-recessive hereditary disorder of the urea cycle caused by a deficiency of the enzyme arginase, which hydrolyses arginine to ornithine and urea. The serum levels of arginine, homoarginine and other guanidino compounds are much higher in hyperargininemic patients than the normal range (Terheggen et al. 1975). Of note, patients suffering from this disorder all have multiple clinical symptoms, such as vomiting, irritability, lethargy, seizures, convulsions, intellectual impairment, spasticity and coma (Marescau et al.1985; Mizutani et al. 1987; Hiramatsu 2003; Deignan et al. 2008, 2010).
The brain as a target of hArg
The severe neurological and cognitive problems observed in hyperargininemia patients and in a mouse model of the disease (arginase deficient mice, Deignan et al. 2008; Lee et al. 2013) as a consequence of excess hArg and other guanidino compounds on the one hand, and impaired cerebrovascular function resulting from hArg deficiency (Pilz et al. 2011; Atzler et al. 2015) on the other hand, point to the CNS as an important target for hArg action. Unfortunately, information about the origin and the functions of hArg in the normal and diseased brain is still very limited. The present short review aims at summarizing the current knowledge on this topic. In addition, in part II of this paper we will present some recent immunohistochemical findings from our laboratory that might help to answer one or the other open question in this context.
HArg levels in the brain, non-neural tissues and bodily fluids
HArg is detectable in numerous tissues and bodily fluids. An analysis of hArg concentrations in some tissues of different mammalian species (Marescau et al. 1992; May et al. 2015) revealed that (1) serum hArg content is highest in fasting human subjects and rats (and lowest in ferrets) and, more importantly for our considerations, (2) hArg content of brain material is in the range of tissues with “established” local hArg synthesis (i.e., of kidney and liver, see below; Table 1). In addition, it was shown by Shiraga et al. (1991) that hArg serum levels were significantly lower in women than in men.
Moreover, in a recent paper (Deignan et al. 2010) findings were summarized on hArg levels in serum, cerebrospinal fluid (CSF) and different regions of post-mortem human brain of nonargininemic human subjects (children, adolescent and adult). Remarkably, brain tissue levels of hArg were estimated to be threefold to fivefold higher than those measurable in the CSF, with the highest concentrations being found in the cerebellar cortex (see Table 2).
Interestingly, Deignan et al. (2010) also found that brain hArg concentrations are in hyperargininergic patients 50–85 % higher than the upper normal values in adults.
HArg in the CNS: substance uptake or local synthesis, or both?
The tissue content of any chemical compound may either result from uptake of the substance itself by the tissue from outside the organ, or from local tissue synthesis. This holds also true for hArg.
Arguments for possible hArg transport into the brain
HArg is mainly carried by cationic amino acid transporters
Forty years ago it was reported that hArg is taken up by slices prepared from human liver (an organ, which is able to synthesize hArg by itself) via two different transport systems, whereby one of them, the “low concentration system”, was only active in patients with lysinuric protein intolerance but not in control cases (Simell and Perheentupa 1974). Two hArg transporters with very similar properties were subsequently found in granulocytes by the same research group (Simell 1975). Later on, however, it became obvious that hArg is mainly (if not exclusively) transported by cationic amino acid transporters (together designated as System y+ by White et al. (1982), which are kinetically distinct from systems transporting neutral and acidic amino acids. The uptake of cationic substrates (including hArg) by these transporters is sodium-independent, stereoselective, pH-insensitive and inhibitable by neutral amino acids in the presence of Na+. Uptake and exodus of System y+ substrates are stimulated by cationic amino acids inside and outside the cell (White and Christensen 1982; White et al. 1982; van Winkle et al. 1985). These cationic amino acid transporters (CATs; Kakuda and MaLoed 1994; Closs et al. 1997) constitute a subfamily of solute carrier family 7 (SLC7), consisting of four closely related transport proteins: CAT1 (SLC7A1), CAT2 (SLC7A2A), CAT2B (SLC7A2B) and CAT3 (SLC7A3) (Closs et al. 2006). Although there is recent evidence for an intracellular hArg transport by another member of the solute carrier family (namely SLC25A29, which is a member of the mitochondrial carrier family; Porcelli et al. 2014), CATs remain the favorite candidate carrier proteins for hArg transport.
Cationic amino acid transporters are highly expressed in different brain cells
An indispensable prerequisite for an uptake/transport of hArg into, or within, the CNS would be the presence of CATs in the brain. With regard to hArg transport into the brain, there are three possible routes: as any other compound, hArg may (1) originate from the blood, being transported to brain tissue through the blood–brain barrier, (2) enter the brain passing the vasculature of circumventricular organs or (3) come from the cerebral spinal fluid transported through the choroid plexus ependymal cells (O´Kane et al. 2006; Jäger et al. 2013; Bernstein et al. 2014). Of note, although closely related, individual members of the CAT family considerably differ with regard to substrate affinity and sensitivity. To our knowledge, it is yet not known if hArg is exclusively or preferentially transported by one or more CAT family members. Hence, the brain expression of all members of CAT family should be taken into consideration. CAT family members show a wide but uneven distribution in the brain, having a transporter-specific (though partly overlapping) expression patterns (Braissant et al. 2001a; Jäger et al. 2013; results from this study). CAT1 has been detected in neurons, astrocytes, oligodendrocytes, while CAT2B is expressed in neurons, oligodendrocytes but not in astrocytes. Last, CAT3, previously regarded as “neuron-specific” transporter (Braissant et al. 2001a; Manner et al. 2003), was recently also found in astrocytes and oligodendrocytes (at least in humans, Jäger et al. 2013; this study). Of note, in humans all three CAT transporters are highly expressed in choroid plexus epithelia, in ependymal cells facing the ventricles and in circumventricular organs (CVOs, with the most abundant expression found in the pineal gland), while hCAT1 and, to a lesser extent, hCAT3 showed a weak to moderate expression in some capillaries in regions with a blood–brain barrier (Jäger et al. 2013; this study). Thus, from a neuroanatomical viewpoint, an uptake of hArg from the blood is conceivable, with CAT1 acting as the most probable transporter through capillaries with an intact blood–brain barrier and any of the CATs in CVO regions. Regarding CSF, the situation is unclear: although all three CATs are abundantly expressed in choroid plexus epithelial and ependymal cells, are the CSF levels of hArg much lower than those in the brain tissue (Deignan et al. 2010)? Hence, the uptake of hArg from the cerebral spinal fluid into the brain via the choroid plexus epithelial and/or ependymal cells would have to work against a concentration gradient. Further studies are needed to clarify if this transportation route does really exist. hArg transport in the opposite direction (i.e., from the brain into the CSF) is more plausible, however. This might be a transportation route to remove hArg from brain tissue. Alternatively, cerebral hArg might be inactivated by degradation within the brain. Arginase, the enzyme controlling hArg degradation and ornithine synthesis from arginine (Jaźwińska-Kozuba et al. 2013) has been detected in the brain. However, it is not clear if this pathway is active in the normal, non-inflamed brain, since there is little evidence for an expression of this enzyme outside activated microglia (Wiesinger 2001; Lisi et al. 2014). Recent findings on a wide distribution of arginase in rat brain neurons (Peters et al. 2013) need to be replicated by others.
Arguments for possible hArg synthesis in the brain
Ornithine transcarbamylase and l-arginine:glycine amidinotransferase are able to synthesize hArg
De novo hArg synthesis has been shown to occur mainly in liver and kidney (Ryan et al. 1968, 1969). The key enzyme for hArg synthesis is ornithine transcarbamylase (OTC; EC 2.1.3.3). Although this enzyme has a higher affinity to ornithine, it also catalyzes the transaminidation reaction of lysine to homocitrulline, thereby initiating hArg formation through the metabolic intermediate homoargininosuccinate (discussed by März et al. 2010). Besides, hArg is generated when lysine substitutes for glycine as a substrate of the enzyme L-arginine:glycine amidinotransferase (AGAT, EC 2.1.4.1; for overview see Jaźwińska-Kozuba et al. 2013 and others), the primary synthesis product of which is creatine (Braissant et al. 2007). Recent evidence suggests that AGAT may be even more important than OTC for the regulation of hArg levels in serum and other tissues (Choe et al. 2013; Kleber et al. 2013). In addition to renal and liver tissue lymphoblasts, pancreatic cells and cardiomyocytes have been shown to be capable of synthesizing hArg from arginine and lysine due to their expression of AGAT (Watanabe et al. 1988; Davids et al. 2012; Kayacelebi et al. 2014).
OTC and AGAT are present in the CNS
HArg synthesis in the brain would assume the presence of either OTC or AGAT in nervous tissue. Relatively little is currently known about the expression of OTC in the CNS. According to gene network data, considerable amounts of OTC mRNA are detectable in different regions of rodent and human brain (for details see data from genenetwork/org dataset 1). Moreover, in a recent work (Lopes-Marques et al. 2012) evidence was provided for the expression of an additional (brain specific?) larger enzyme transcript (namely OTC-t3) in human brain samples. However, the expression of OTC protein in nervous tissue is largely unexplored (with the exception of one paper describing OTC in brain capillaries of Alzheimer disease (AD) patients, but not at all in controls brains; Bensemain et al. 2009). We, therefore, felt encouraged to undertake a histochemical mapping study of the human brain with regard to the regional distribution and cellular localization of OTC (see experimental work herein). Using well-characterized, monospecific antibodies we could reveal that OTC is widely distributed throughout adult human non-AD brains, being expressed in numerous gray and white matter neurons, oligodendrocytes, ependymal cells, choroid plexus epithelial cells and some blood vessels. Thus, our findings apparently contradict those published by Bensemain et al. (2009), but are in good accordance with mRNA data (genenetwork.org dataset 2). Concerning the cerebral appearance of AGAT, comprehensive work from Braissant´s group (Braissant et al. 2001b, 2005a, b, 2007) and others (Wyss and Kaddurah-Daouk 2000; Cullen et al. 2006) has demonstrated a wide distribution pattern of both AGAT mRNA and protein in the rat brain, with AGAT protein expression being found in every cell type (reviewed in detail in Braissant et al. 2007). In the current study we could largely replicate the protein pattern for rat brain AGAT known from work of others. However, since nearly no data are available for human brain AGAT expression, we in this study have performed immunostaining of human brain tissue. We found a distribution pattern, which was slightly different from those observed in rats. This is particularly applicable to the occurrence of single gray and white matter neurons with extremely intense intracellular AGAT staining.
In sum, although direct evidence for cerebral hArg synthesis is lacking, it can be stated that the enzyme machinery for hArg synthesis is abundantly expressed in the mammalian brain. Whether uptake or synthesis (or a combination of both) is responsible for brain hArg levels remains unclear and warrants further attention.
HArg and brain function
Putative roles of hArg in the healthy brain
The functional importance of hArg for the healthy brain is poorly understood. Results of physiological, biochemical and pharmacological experiments have shown a variety of different effects of hArg on CNS processes, including its ability to modify the EEG of rats (Yokoi et al. 1984–1985), to act as a substrate of nNOS (Lambert et al. 1992; Yokoi et al. 1994), to block lysine transport in the brain (Tews and Harper 1983), to inhibit the uptake of L-[3H]arginine into rat brain synaptosomes (Aldridge and Collard 1996), to decrease GABA responses to pentylenetetrazol-induced GABA responses on mouse neurons in cell culture (De Deyn et al. 1990), to stimulate the activity of the rat brain enzymes acetylcholine esterase (Delwing-de Lima et al. 2010) and ecto-nucleoside triphosphate diphosphohydrolase 1 (Balz et al. 2003) and to alter voluntary feed intake in chickens (Angkanaporn et al. 1987). In addition, cyclic dipeptide cyclo[Tyr(et)]-homoarginine was shown to exert antinociceptive effects in rats when administered into the lateral ventricles (Sato et al. 1984). However, whether hArg does really play or not a significant role in normal brain function remains to be elucidated.
With regard to brain diseases, evidence has accumulated in favor of an involvement of hArg in some of them. Remarkably, both hArg excess and deficiency may contribute to CNS-related disorders.
Possible roles of hArg in the diseased brain
HArg excess and brain disorders
As aforementioned, do hyperargininemic patients show severe neurological signs, which are most probably evoked by increased concentrations of hArg and other guanidino compounds in the brain (Deignan et al. 2010)? Of these neurological symptoms, only the putative implication of hArg for convulsions/seizures has been studied in some detail (Yokoi et al. 1984–1985; Shiraga et al. 1991; Hiramatsu 2003). It is well known that homoarginine and other guanidino compounds are able to induce epileptic seizures when administered intracisternally to experimental animals. Moreover, it has been shown that endogenous guanidino compound levels change before and after seizures in the brain of epileptic animals (reviewed in Shiraga et al. 1991; Hiramatsu 2003). Interestingly, serum hArg levels of the male patients are significantly lower (not higher as one would expect) in epileptic neurological patients with symptomatic generalized epilepsy than in non-epileptic patients (Shiraga et al. 1991). This observation, together with numerous clinical data, supports the notion that not so much elevated cerebral levels of hArg but increased concentrations of other endogenous guanidino compounds evoke seizures in patients. There is some evidence that the accumulation of guanidinoacetic acid in brain and bodily fluids is mainly responsible for intractable seizures and the movement disorder, both exclusively found in conditions of guanidinoacetate methyltransferase (GAMT) deficiency (Schulze 2003). This enzyme catalyzes the formation of creatine from guaninioacetatic acid. Its absence leads to the accumulation of guaninoacetate and at the same time to creatine deficiency. Having autosomal-recessive traits, GAMT deficiency typically presents with seizures during infancy and childhood alongside with other neurological signs (i.e., muscular hypotonia, global developmental delay and extrapyramidal symptoms; Vodopiutz et al. 2007; Gordon 2010). In the endeavor to provide a meaningful explanation for the convulsion/seizure-inducing activity of hArg and other guanidino compounds, Hiramatsu (2003) has speculated about putative roles of these substances in the brain (influences on membrane fluidity, NMDA receptor function, generation of free radicals, energy production and protection of neurons from apoptosis and cell death). Future investigations will have to establish which of the possible mechanisms is in fact responsible for guaninino compound-induced seizures. Another known complication in hyperargininemia is cognitive impairment, pointing to a possible role of hArg in processes memory storage and learning. Unfortunately, to our knowledge there are no studies dealing with this topic (except two papers showing that one the hArg-synthesizing enzymes, OTC is up-regulated in brains of AD patients, which might, theoretically, might lead to increased cerebral levels of hArg; Bensemain et al. 2009; Hansmannel et al. 2009). Last but not least, it should be taken into account that hArg may be neurotoxic. When hArg is taken up in great amounts with the food, it contributes to the development of a severe neurological disease, called neurolathyrism, which was a serious health problem in some countries (Ethiopia, India, Bangladesh, Afghanistan) until recently. Neurolathyrism is associated with a complex pattern of alterations in the glutamatergic neurotransmitter system of the motor region of brain cortex. It is a neurological disorder resulting from excessive consumption of Grass pea (Lathyrus sativus). Grass pea contains, among other toxic compounds (such as the amino acid β-N-oxalyl-l-α, β-diaminopropionic acid), high levels of hArg, which is a substrate of NO production. The consequence is a drastically increased NO formation in the CNS, which in its turn stimulates the formation of peroxynitrite radicals. These radicals cause irreparable damage to mitochondria and other cellular elements leading to motor neuron degeneration (for overview see Khandare et al. 2013).
HArg deficiency and brain disorders
HArg is inversely associated with subclinical vascular disease and with risk for cardiovascular disease events. In patients with AGAT deficiency intracellular energy stores are reduced (ATP and phosphocreatine). These patients show profound hArg deficiency, which has been linked to an improved metabolic risk profile on the one hand, but to impaired cardiac and cerebrovascular function on the other hand (Atzler et al. 2014, 2015). Similar results were obtained with AGAT deficient mice. However, research is this field has just begun, and more experiments are necessary to clarify if hArg is just a suitable marker or a causal mediator in cerebrovascular disease (Atzler et al. 2015). A strong down-regulation of the hArg synthesizing enzyme, AGAT, was found in mood-disordered suicide completers, which might be a first hint for a possible role of decreased hArg in the pathophysiology of neuropsychiatric disorders (Fiori et al. 2011). Surely, this conjecture remains a speculation until measurements will really show changes in brain hArg concentrations in postmortem brains of depressives.
In conclusion it can be said that balanced cerebral hArg levels are obviously mandatory to keep the brain healthy.
Part II: Own experimental data
The paucity of information about the possible origin of cerebral hArg inspired us to immunolocalize the transporter proteins hCAT1, hCAT2 and hCAT3 as well as the hArg synthesizing enzymes for OTC and AGAT in human brain and AGAT in rat brain.
Materials and methods
Subjects
Human postmortem brains were obtained from the New Magdeburg brain collection. The case recruitment, acquisition of personal data, performance of autopsy and handling of autoptic material were conducted in accordance with the Declaration of Helsinki and was approved by the Ethical Committee of Magdeburg. The brains of five human subjects (three males, two females; mean age: 51.4 ± 2.8 years) without a history of neuropsychiatric disorder were investigated. None of the subjects had a history of substance abuse or alcoholism. An experienced neuropathologist ruled out changes due to neurodegenerative or traumatic processes.
Tissue processing
The adult subjects’ brains were removed within 9–21 h after death and fixed in toto in 8 % phosphate-buffered formaldehyde for at least 2 months. The frontal and occipital poles were separated by coronal sectioning anterior to the genu and posterior to the splenium of the corpus callosum. After embedding in paraffin, serial coronal sections of the middle block were cut (20 μm) and mounted. The distance between the sections was 1 mm. Every 50th section was Nissl- and myelin-stained as described earlier (Bernstein et al. 1998).
Human brain immunohistochemistry
Antisera
To immunolocalize the cationic amino acid transporters hCAT1, hCAT2 and hCAT3 we used monospecific polyclonal antisera as recently described (Jäger et al. 2013). HCAT1 was detected by employing two different antisera. One was manufactured by immunizing rabbits with the designed appropriate synthetic peptide (anti-human CAT1-IgG; DPC Biermann GmbH, Bad Nauheim, Germany). The second anti-hCAT1-antibody was a commercially available, affinity purified IgG antiserum raised in rabbits (SLC7A1, Biozol Eching, Germany). Both anti-hCAT1 antisera yielded equally good and specific staining results. Immunodetection of HCAT2 was performed using a rabbit polyclonal antiserum generated against the C-terminal domain of the human origin peptide (sc 87036, Santa Cruz Biotechnology, USA). Immunolocalization of hCAT3 was carried out using a rabbit polyclonal antiserum produced against the synthetic human peptide (SLC7A3, Biozol Eching, Germany).
To immunolocalize l-ornithine transcarbamoylase we used a monospecific polyclonal anti-OTC antiserum generated in rabbits against synthetic peptide corresponding to human OTC aa 71–98, conjugated to keyhole limpet hemocyanin (ab91418 from Abcam). For the immunodetection of l-arginine:glycine amidinotransferase a monospecific polyclonal anti-AGAT antiserum was employed, which was produced in rabbits against the synthetic peptide 101–115 of the human AGAT (SAB1101113 from Sigma-Aldrich). The dilutions for AGAT and OTC antisera were 1:200 in phosphate-buffered saline.
Whole brain frontal sections were collected at intervals of about 1.8 cm from the level 2 cm rostral to the splenium to the posterior splenium and from the central portion of the raphe nuclei to the central portion of the olivary nuclei. After dewaxing, the sections were boiled in 10 mM citrate buffer (pH = 6.0) and then pre-incubated with methanol/H2O2 to suppress endogenous peroxidases. After repeated washing with phosphate-buffered saline (PBS), the respective antibodies (anti-hCAT 1–3, OTC or AGAT) were applied at dilutions of 1:200 in PBS. For visualization, the avidin–biotin method (Vectastain-peroxidase kit) with 3, 3′-diaminobenzidine as chromogen was used. The color reaction was enhanced by adding 2 ml of a 0.5 % nickel ammonium sulfate solution to the diaminobenzidine as described previously (Bernstein et al. 1999). The procedure yielded a dark purplish-blue to dark-blue color reaction product. For negative controls, the primary antibodies were replaced with buffer or normal serum. No immunostaining was found in control sections. As a positive reference tissue for OTC and AGAT immunolocalizations human liver was used.
Rat brain histology and immunohistochemistry
For all procedures ethical approval was sought according to the National Act on the Use of Experimental Animals (Germany). At postnatal day 56, rats were killed. Animals (N = 7) were anesthetized with chloral hydrate and transcardially perfused with 8 % formalin. Brains were removed from the cranium, fixed in formalin and then embedded in paraffin. Serial sections (6 µm thick) were cut. Every tenth section was stained for Nissl. For immunohistochemistry we used a monospecific polyclonal antiserum manufactured against AGAT. The immunohistochemical staining protocol and controls were as for human brains.
Results
HCAT immunoreactivity
In this study we could replicate our previous findings (Jäger et al. 2013) on a wide regional and cellular distribution of cationic amino acid transporters in human brain (hCATs). All three hCAT1s were predominantly localized in neurons (Fig. 1a, b), but were also expressed in numerous astrocytes, oligodendrocytes and choroid plexus epithelial cells (Fig. 1c–f). HCAT1 and hCAT3 were in addition observed in some small blood vessels (Fig. 1b, g). The highest density of hCAT-expressing neurons was found in the hypothalamus, in some areas of the cerebral cortex, the thalamic reticular nucleus and the caudate nucleus, whereas weak to moderate expression was detected in the hippocampus, the prefrontal cortex, pontine neurons, brain stem and cerebellum. In contrast to what has been found in rodent brain by others (Braissant et al. 2001a; Manner et al. 2003), we detected hCAT2 and hCAT3 also in a subpopulation of human brain astrocytes. Remarkably, the pineal gland (one of the CVOs) stood out by very intense immunostaining for all three hCATs (Fig. 1e).

Cellular localization of hCAT1, hCAT2 and hCAT3 in the adult human brain. a HCAT1 expressing pyramidal cells and interneurons in the hippocampus. Bar 40 µm. b HCAT3 immunoreactive layer IV interneurons and an immunoreactive capillary in the prefrontal cortex. Bar 30 µm. c HCAT1 immunopositive astrocyte in the hippocampus at higher magnification. Bar 15 µm. d HCAT2 immunoreactive astrocytes in the temporal cortex. Bar 40 µm. e Strong expression of hCAT1in the pineal gland. Bar 50 µm. f Expression of hCAT3 in the choroid plexus. Bar 40 µm. g HCAT1 immunoreactive blood vessels. Bar 40 µm. h Control reaction. After replacement of hCAT3 antiserum by normal rabbit serum no specific stainings are visible. Bar 40 µm
OTC immunoreactivity in human brain
OTC enzyme was immunohistochemically localized to multiple neurons situated in all cortical areas, thalamus, hippocampus, amygdala, hypothalamus, cerebellum and brain stem (Fig. 2a–d). The intraneuronal immunostaining was restricted to the cytoplasm and some dendrites. No immunoreaction appeared in neuronal nuclei. Besides, OTC was revealed in white matter interstitial neurons, and to a lesser extent, in oligodendrocytes, ependymal cells facing the lateral ventricles, choroid plexus epithelial cells and some blood vessels.

Immunolocalization of OTC and AGAT in human and rat brain. a Neurons in the dorso-lateral prefrontal cortex immunostained for OTC. Bar 40 µm. b OTC expressing thalamic neurons. Bar 75 µm. c Cellular OTC distribution in the cerebellum. Both Purkinje cells and granular cells exhibit OTC immunoreactivity. Bar 40 µm. d Pontine neurons are strongly immunostained for OTC. Bar 30 µm. e A single human brain prefrontal neuron showing an intense immunostaining for AGAT. A vast majority of cortical neurons are only weakly to moderately stained for this enzyme protein. Bar 40 µm. f AGAT immunoreactive fibers forming fine networks can be observed in the cortical white matter (this microphotograph) as well as in many other brain areas of the human brain. Bar 25 µm. g AGAT immunoreactive perirhinal cortex neurons in the rat brain. Bar 40 µm. h Abundant expression of AGAT in human liver cells. Bar 50 µm
AGAT immunoreactivity in human brain
In the human brain, AGAT showed a remarkable distribution in that numerous white matter interstitial neurons and a small number of gray matter cortical neurons stood out by a very intense immunostaining (Fig. 2e). In addition, moderate immunostaining was found in many cortical and hypothalamic neurons as well as in cerebellar Purkinje cells. Furthermore, a fine network of AGAT-immunoreactive fibers was seen in many cortical gray and white matter areas (Fig. 2f). Extraneuronally, AGAT immunoreactivity was identified in some white matter oligodendrocytes, choroid plexus epithelial cells and small blood vessels.
AGAT immunoreactivity in rat brain
AGAT immunoreactive material was seen in many cortical (Fig. 2g) and subcortical neurons, oligodendrocytes, choroid plexus epithelium and blood vessels. As a positive reference tissue we used human liver, where AGAT was found in most (if not all) hepatocytes (Fig. 2h).
Conclusions
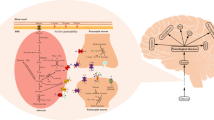
The immunohistochemical demonstration of the presence of hCAt1, hCAt2 and hCAT3 as well as of OTC and AGAT in human and rat CNS lends support to the idea that cerebral hArg may originate both from uptake and local synthesis (Fig. 3). For normal brain function hArg should be in balance. Both hArg excess and deficiency may lead to characteristic brain pathologic and mental alterations (Fig. 4).

Hypothetical dual origin of cerebral hArg. HArg may be taken up by the brain, transferred in the brain and transported out of the brain by cationic amino acid transporters (CATs). In addition, hArg may be synthesized by OTC and/or AGAT in nervous tissue. A degradation of hARG by brain arginase is questionable, however

Schematic drawing summarizing some brain pathologic alterations evoked by either hArg excess or deficit
Abbreviations
- AGAT:
-
l-Arginineglycine amidinotransferase
- CAT:
-
Cationic amino acid transporter
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CVO:
-
Circumventricular organs
- DL:
-
Detection limit
- EEG:
-
Electroencephalogramm
- GABA:
-
γ-Aminobutyric acid
- GAMT:
-
Guanidinoacetate methyltransferase
- hArg:
-
l-Homoarginine
- hCAT:
-
Human cationic amino acid transporter
- IgG:
-
Immunoglobulin G
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- eNOS:
-
Endothelial nitric oxide synthase
- iNOS:
-
Inducible nitric oxide synthase
- nNOS:
-
Neuronal nitric oxide synthase
- OTC:
-
Ornithine transcarbamylase
- SLC:
-
Solute carriers (family of transporter proteins)
- Tyr:
-
Tyrosine
References
Aldridge CR, Collard KJ (1996) The characteristics of arginine transport by rat cerebellar and cortical synaptosomes. Neurochem Res 21:1539–1546
Angkanaporn K, Ravindran V, Mollah Y, Bryden WL (1987) Homoarginine influences voluntary feed intake, tissue basic amino acid concentrations and arginase activity in chickens. J Nutr 127:1128–1136
Atzler D, Gore MO, Ayers CR, Choe CU, Böger RH, de Lemos JA, McGuire DK, Schwedhelm E (2014) Homoarginine and cardiovascular outcome in the population-based Dallas Heart Study. Arterioscler Thromb Vasc Biol 34:2501–2507
Atzler D, Schwedhelm E, Choe CU (2015) l-Homoarginine and cardiovascular disease. Curr Opin Clin Nutr Metab Care 18:83–88
Balz D, de Souza Wyse AT, Morsch VM, da Silva AC, Vieira VL, Morsch AL, Schetinger MR (2003) In vitro effects of l-arginine and guanidino compounds on NTPDase1 and 5′-nucleotidase activities from rat brain synaptosomes. Int J Dev Neurosci 21:75–82
Bensemain F, Hot D, Ferreira S, Dumont J, Bombois S, Maurage CA, Huot L, Hermant X, Levillain E, Hubans C, Hansmannel F, Chapuis J, Hauw JJ, Schraen S, Lemoine Y, Buée L, Berr C, Mann D, Pasquier F, Amouyel P, Lambert JC (2009) Evidence for induction of the ornithine transcarbamylase expression in Alzheimer’s disease. Mol Psychiatry 14:106–116
Bernstein HG, Stanarius A, Baumann B, Henning H, Krell D, Danos P, Falkai P, Bogerts B (1998) Nitric oxide synthase-containing neurons in the human hypothalamus: reduced number of immunoreactive cells in the paraventricular nucleus of depressive patients and schizophrenics. Neuroscience 83:867–875
Bernstein HG, Baumann B, Danos P, Diekmann S, Bogerts B, Gundelfinger ED, Braunewell KH (1999) Regional and cellular distribution of neural visinin-like protein immunoreactivities (VILIP-1 and VILIP-3) in human brain. J Neurocytol 28:655–662
Bernstein HG, Hölzl G, Dobrowolny H, Hildebrandt J, Trübner K, Krohn M, Bogerts B, Pahnke J (2014) Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech Ageing Dev 141:12–21
Braissant O, Gotoh T, Loup M, Mori M, Bachmann C (2001a) Differential expression of the cationic amino acid transporter 2(B) in the adult rat brain. Brain Res Mol Brain Res 91:189–195
Braissant O, Henry H, Loup M, Eilers B, Bachmann C (2001b) Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Mol Brain Res 86:193–201
Braissant O, Villard A, Henry H, Speer O, Wallimann T, Bachmann C (2005a) Synthesis and transport of creatine in the central nervous system. In: Clinical and molecular aspects of defects in creatine and polyol metabolism. Jakobs C, Stöckler-Ipsiroglu eds. (SPS Verlagsgesellschaft Heilbronn, Germany), pp 49–63
Braissant O, Henry H, Villard AM, Speer O, Wallimann T, Bachmann C (2005b) Creatine synthesis and transport during rat embryogenesis.: spatiotemporal expression of AGAT, GAMT and CT1. BMC Dev Biol 5:9
Braissant O, Bachmann C, Henry H (2007) Expression and function of AGAT, GAMT and CT1 in the mammalian brain. Subcell Biochem 46:67–81
Choe CU, Atzler D, Wild PS, Carter AM, Böger RH, Ojeda F, Simova O, Stockebrand M, Lackner K, Nabuurs C, Marescau B, Streichert T, Müller C, Lüneburg N, De Deyn PP, Benndorf RA, Baldus S, Gerloff C, Blankenberg S, Heerschap A, Grant PJ, Magnus T, Zeller T, Isbrandt D, Schwedhelm E (2013) Homoarginine levels are regulated by l-arginine:glycine amidinotransferase and affect stroke outcome: results from human and murine studies. Circulation 128:1451–1461
Closs EI, Gräf P, Habermeier A, Cunningham JM, Förstermann U (1997) Human cationic amino acid transporters hCAT-1, hCAT-2A and hCAT-2B: three related carriers with distinct transport properties. Biochemistry 36:6462–6468
Closs EI, Boissel JP, Habermeier A, Rotmann A (2006) Structure and function of cationic amino acid transporters (CATs). J Membr Biol 213:67–77
Cullen ME, Yuen AH, Felkin LE, Smolenski RT, Hall JL, Grindle S, Miller LW, Birks EJ, Yacoub MH, Barton PJ (2006) Myocardial expression of the arginine: glycine amidinotransferase gene is elevated in heart failure and normalized after recovery: potential implications for local creatine synthesis. Circulation 114(Suppl I):16–20
Davids M, Ndika JD, Salomons GS, Blom HJ, Teerlink T (2012) Promiscuous activity of arginine:glycine amidinotransferase is responsible for the synthesis of the novel cardiovascular risk factor homoarginine. FEBS Lett 586:3653–3657
De Deyn PP, Marescau B, MacDonald RL (1990) Epilepsy and the GABA-hypothesis a brief review and some examples. Acta Neurol Belg 90:65–81
Deignan JL, Marescau B, Livesay JC, Iyer RK, De Deyn PP, Cederbaum SD, Grody WW (2008) Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Mol Genet Metab 93:172–178
Deignan JL, De Deyn PP, Cederbaum SD, Fuchshuber A, Roth B, Gsell W, Marescau B (2010) Guanidino compound levels in blood, cerebrospinal fluid, and post-mortem brain material of petients with argininemia. Mol Genet Metab 100:531–536
Delwing-de Lima D, Wollinger LF, Casagrande AC, Delwing F, da Cruz JG, Wyse AT, Delwing-Dal Magro D (2010) Guanidino compounds inhibit acetylcholinesterase and butyrylcholinesterase activities: effect neuroprotector of vitamins E plus C. Int J Dev Neurosci 28:465–473
Fiori LM, Bureau A, Labbe A, Croteau J, Noël S, Mérette C, Turecki G (2011) Global gene expression profiling of the polyamine system in suicide completers. Int J Neuropsychopharmacol 14:595–605
Fishman WH, Sie HG (1971) Organ-specific inhibition of human alkaline phosphatase isoenzymes of liver, bone, intestine and placenta; l-phenylalanine, l-tryptophan and L homoarginine. Enzymologia 41:141–167
Genenetwork.org dataset 1. http://www.genenetwork.org/webqtl/WebQTL.py?cmd=search&gene=Otc
Genenetwork.org dataset 2. http://www.genenetwork.org/webqtl/WebQTL.py?cmd=search&gene=Otc
Gordon N (2010) Guanidinoacetate methyltransferase deficiency (GAMT). Brain Dev 32:79–81
Hansmannel F, Lendon C, Pasquier F, Dumont J, Hannequin D, Chapuis J, Laumet G, Ayral AM, Galimberti D, Scarpini E, Campion D, Amouyel P, Lambert JC (2009) Is the ornithine transcarbamylase gene a genetic determinant of Alzheimer’s disease? Neurosci Lett 449:76–80
Hiramatsu M (2003) A role for guanidine compounds in the brain. Mol Cell Biochem 244:57–62
Jäger K, Wolf S, Dobrowolny H, Steiner J, Nave H, Maronde E, Bogerts B, Bernstein HG (2013) Differential topochemistry of three cationic amino acid transporter proteins, hCAT1, hCAT2 and hCAT3, in the adult human brain. Amino Acids 44:423–433
Jaźwińska-Kozuba A, Martens-Lobenhoffer J, Kruszelnicka O, Rycaj J, Chyrchel B, Surdacki A, Bode-Böger SM (2013) Opposite associations of plasma homoarginine and ornithine with arginine in healthy children and adolescents. Int J Mol Sci 14:21819–21832
Kakuda DK, MaLoed CL (1994) Na+-independent transport (uniport) of amino acids and glucose in mammalian cells. J Exp Biol 196:93–108
Kayacelebi AA, Nguyen TH, Neil C, Horowitz JD, Jordan J, Tsikas D (2014) Homoarginine and 3-nitrotyrosine in patients with takotsubo cardiomyopathy. Int J Cardiol 173:546–547
Khandare AL, Ankulu M, Aparna N (2013) Role of glutamate and nitric oxide in onset of motor neuron degeneration in neurolathyrism. Neurotoxicology 34(269–274):21
Kikuchi Y, Takagi M, Parmley RT, Ghanta VK, Hiramoto RN (1982) Inhibitory effect of l-homoarginine on murine osteosarcoma cell proliferation. Cancer Res 42:1072–1077
Kleber ME, Seppälä I, Pilz S, Hoffmann MM, Tomaschitz A, Oksala N, Raitoharju E, Lyytikäinen LP, Mäkelä KM, Laaksonen R, Kähönen M, Raitakari OT, Huang J, Kienreich K, Fahrleitner-Pammer A, Drechsler C, Krane V, Boehm BO, Koenig W, Wanner C, Lehtimäki T, März W, Meinitzer A (2013) Genome-wide association study identifies 3 genomic loci significantly associated with serum levels of homoarginine: the AtheroRemo Consortium. Circ Cardiovasc Genet 6:505–513
Lambert LE, French JF, Whitten JP, Baron BM, McDonald IA (1992) Characterization of cell selectivity of two novel inhibitors of nitric oxide synthesis. Eur J Pharmacol 216:131–134
Lee EK, Hu C, Bhargava R, Ponnusamy R, Park H, Novicoff S, Rozengurt N, Marescau B, De Deyn P, Stout D, Schlichting L, Grody WW, Cederbaum SD, Lipshutz GS (2013) AAV-based gene therapy prevents neuropathology and results in normal cognitive development in the hyperargininemic mouse. Gene Ther 20:785–796
Lisi L, Tramutola A, Navarra P, Dello Russo C (2014) Antiretroviral agents increase NO production in gp120/IFNγ-stimulated cultures of rat microglia via an arginase-dependent mechanism. J Neuroimmunol 266:24–32
Lopes-Marques M, Pereira-Castro I, Amorim A, Azevedo L (2012) Characterization of the human orhnithine transcaramylase 3′ untranslated regulatory region. DNA Cell Biol 31:427–433
Manner CK, Nicholson B, MacLeod CL (2003) CAT2 arginine transporter deficiency significantly reduces iNOS-mediated NO production in astrocytes. J Neurochem 85:476–482
Marescau B, Qureshi IA, De Deyn P, Letarte J, Ryba R, Lowenthal A (1985) Guanidino compounds in plasma, urine and cerebrospinal fluid of hyperargininemic patients during therapy. Clin Chim Acta 146:21–27
Marescau B, Deshmukh DR, Kockx M, Possemiers I, Qureshi IA, Wiechert P, De Deyn PP (1992) Guanidino compounds in serum, urine, liver, kidney, and brain of man and some ureotelic animals. Metabolism 41:526–532
März W, Meinitzer A, Drechsler C, Pilz S, Krane V, Kleber ME, Fischer J, Winkelmann BR, Böhm BO, Ritz E, Wanner C (2010) Homoarginine, cardiovascular risk, and mortality. Circulation 122:967–975
May M, Kayacelebi AA, Batkai S, Jordan J, Tsikas D, Engeli S (2015) Plasma and tissue homoarginine concentrations in healthy and obese humans. Amino Acids. doi:10.1007/s00726-015-1922-4 (in press)
Mizutani N, Hayakawa C, Ohya Y, Watanabe K, Watanabe Y, Mori A (1987) Guanidino compounds in hyperargininemia. Tohoku J Exp Med 153:197–205
O´Kane RL, Viña JR, Simpson I, Zaragozá R, Mokashi A, Hawkins RA (2006) Cationic amino acid transport across blood-brain barrier is mediated exclusively by system y+. Am J Physiol Endocrinol Metab 291:E412–E419
Peters D, Berger J, Langnaese K, Derst C, Madai VI, Krauss M, Fischer KD, Veh RW, Laube G (2010) Arginase and arginine decarboxylase - where do the putative gate keepers of polyamine synthesis reside in the rat brain? PlosOne 8:e66735
Pilz S, Meinitzer A, Tomaschitz A, Drechsler C, Ritz E, Krane V, Wanner C, Boehm BO, März W (2011) Low homoarginine concentration is a novel risk factor for heart disease. Heart 97:1222–1227
Pilz S, Meinitzer A, Tomaschitz A, Kienreich K, Fahrleitner-Pammer A, Drechsler C, Boehm BO, März W (2012) Homoarginine deficiency is associated with increased bone turnover. Osteoporos Int 23:2731–2732
Porcelli V, Fiermonte G, Longo A, Palmieri F (2014) The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J Biol Chem 289:13374–13384
Ryan WL, Barak AJ, Johnson RJ (1968) Lysine, homocitrulline, and homoarginine metabolism by the isolated perfused rat liver. Arch Biochem Biophys 123(2):294–297
Ryan WL, Johnson RJ, Dimari S (1969) Homoarginine synthesis by rat kidney. Arch Biochem Biophys 131:521–526
Sato T, Sakurada S, Sakurada T, Kisara K, Sasaki Y, Akutsu Y, Suzuki K (1984) Comparison of the entinociceptive effect beween the cyclic dipeptide cyclo[Tyr(Et)-homoarginine and the linear dipeptide Boc-Tyr(Et)-homoarginine-OMe in rats. Jpn J Pharmacol 34:1–8
Schulze A (2003) Creatine deficiency syndromes. Mol Cell Biochem 244:143–150
Shiraga H, Watanabe Y, Mori A (1991) Guanidino compound levels in the serum of healthy adults and epileptic patients. Epilepsy Res 8:142–148
Simell O (1975) Diamino acid transport into granulocytes and liver slices of patients with lysinuric protein intolerance. Pediatr Res 9:504–508
Simell O, Perheentupa J (1974) Transport of homoarginine into liver slices of patients with lysinuric protein intolerance (LPI). Pediatr Res 8:904
Steib H (1926) Über d, I-α Methylarginin. Hoppe Seyler’s Z Physiol Chem 155:279–291
Stevens CM, Bush JA (1950) New synthesis of α-amino-ε-guanidino-n-caproic acid (homoarginine) and its possible conversion in vivo into lysine. J Biol Chem 183:139–147
Terheggen HG, Lowenthal A, Lavinha F, Colombo JP (1975) Familial hyperargininaemia. Arch Dis Child 50:57–62
Tews JK, Harper AE (1983) Atypical amino acids inhibit histidine, valine, or lysine transport into rat brain. Am J Physiol 245:R556–R563
Van Winkle LJ, Christensen HN, Campione AL (1985) Na+-dependent transport of basic, zwitterionic, and bicyclic amino acids by a broad-scope system in mouse blastocysts. J Biol Chem 260:12118–12123
Vodopiutz J, Item CB, Häusler M, Korall H, Bodamer OA (2007) Severe speech delay as the presenting symptom of guanidinoacetate methyltransferase deficiency. J Child Neurol 22:773–774
Watanabe Y, Yokoi I, Watanabe S, Sugi H, Mori A (1988) Formation of 2-guanidinoethanol by a transamidination reaction from arginine and ethanolamine by the rat kidney and pancreas. Life Sci 43:295–302
White MF, Christensen HN (1982) The two-way flux of cationic amino acids across the plasma membrane of mammalian cells is largely explained by a single transport system. J Biol Chem 257:10069–10080
White MF, Gazzola GC, Christensen HN (1982) Cationic amino acid transport into cultured animal cells, part I. Influx into cultured human fibroblasts. J Biol Chem 257:4443–4449
Wiesinger H (2001) Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog Neurobiol 64:365–391
Wyss M, Kaddurah-Daouk R (2000) Creatine and creatine metabolism. Physiol Rev 80:1107–1213
Yokoi I, Toma J, Mori A (1984–1985) The effect of homoarginine on the EEG of rats. Neurochem Pathol 2:295–300
Yokoi I, Kabuto H, Habu H, Inada K, Toma J, Mori A (1994) Structure-activity relationships of arginine analogues on nitric oxide synthase activity in the rat brain. Neuropharmacology 33:1261–1265
Acknowledgments
The authors wish to thank B. Jerzykiewicz for excellent technical assistance.
Conflict of interest
The authors declare no conflict of interest.
Ethical statement
Brains were obtained in accordance with existing German and European Union regulations from the Magdeburg Brain Bank. All experimental procedures were in addition approved by the Ethical Committee of Magdeburg.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bernstein, HG., Jäger, K., Dobrowolny, H. et al. Possible sources and functions of l-homoarginine in the brain: review of the literature and own findings. Amino Acids 47, 1729–1740 (2015). https://doi.org/10.1007/s00726-015-1960-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-1960-y