
Abstract
Hyperhomocysteinemia induces vascular endothelial dysfunction, an early hallmark of atherogenesis. While higher levels of circulating asymmetric dimethylarginine (ADMA) and symmetric dimethyl arginine (SDMA), endogenous inhibitors of nitric oxide synthesis, have been associated with increased cardiovascular risk, the role that ADMA and SDMA play in the initiation of hyperhomocysteinemia-induced endothelial dysfunction remains still controversial. In the present study, we studied the changes of circulating ADMA and SDMA in a rat model of acutely hyperhomocysteinemia-induced endothelial dysfunction. In healthy rats, endothelium-related vascular reactivity (measured as acetylcholine-induced transient decrease in mean arterial blood pressure), plasma ADMA and SDMA, total plasma homocysteine (tHcy), cysteine and glutathione were measured before and 2, 4 and 6 h after methionine loading or vehicle. mRNA expression of hepatic dimethylarginine dimethylaminohydrolase-1 (DDAH1), a key protein responsible for ADMA metabolism, was measured 6 h after the methionine loading or the vehicle. Expectedly, methionine load induced a sustained increase in tHcy (up to 54.9 ± 1.9 µM) and a 30 % decrease in vascular reactivity compared to the baseline values. Plasma ADMA and SDMA decreased transiently after the methionine load. Hepatic mRNA expression of DDAH1, cathepsin D, and ubiquitin were significantly lower 6 h after the methionine load than after the vehicle. The absence of an elevation of circulating ADMA and SDMA in this model suggests that endothelial dysfunction induced by acute hyperhomocysteinemia cannot be explained by an up-regulation of protein arginine methyltransferases or a down-regulation of DDAH1. In experimental endothelial dysfunction induced by acute hyperhomocysteinemia, down-regulation of the proteasome is likely to dampen the release of ADMA and SDMA in the circulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Elevated total plasma homocysteine (tHcy) is associated with a risk for cardiovascular diseases and diabetes mellitus in the general population (Andreotti et al. 2000; Zylberstein et al. 2004; Lentz and Haynes 2004). Moderate hyperhomocysteinemia (tHcy range 10–30 µM) is an independent predictor of atherothrombotic events (Casas et al. 2005; De Bree et al. 2002). It has been repeatedly demonstrated in murine models and in humans that acute or chronic increase in tHcy, induced by methionine load and/or by folate deficiency, promotes low-grade inflammation and endothelial dysfunction, hallmarks of atherogenesis (Bellamy et al. 1998; Chambers et al. 1999). Thus, several large prospective studies have evaluated the consequences of lowering tHcy in patients with established cardiovascular diseases (Loscalzo 2006). Unexpectedly, all the clinical trials using dietary folate supplementation to efficiently lower elevated tHcy in patients did not show any protective effects on secondary adverse vascular events, suggesting that hyperhomocysteinemia could be an early biomarker of cardiovascular risk rather than an independent risk factor, as first postulated (den Heijer et al. 2007; Jamison et al. 2007). Besides the implication of reactive oxygen species (ROS) production, the mechanisms underlying vascular dysfunction induced by hyperhomocysteinemia are largely debated (Tousoulis et al. 2011; Dayal and Lentz 2008; Jakubowski 2006).
Acute experimental hyperhomocysteinemia induced by the administration of methionine, the precursor of homocysteine (Hcy), has been used as a uniquely available approach to identify the specific effects of Hcy and methionine on vascular functions, independently of other factors associated with chronic hyperhomocysteinemia and/or its modulation by the B vitamins status (Dayal and Lentz 2008; Hanratty et al. 2001). Using this approach, some studies reported a strong correlation between impairment in vascular reactivity and increases in plasma levels of asymmetric dimethylarginine (ADMA), an endogenous inhibitor of NO synthesis (Böger et al. 2000, 2001; Fu et al. 2005; Stuhlinger et al. 2003; Tsikas et al. 2000a). In vitro studies on endothelial cells have further demonstrated that increased levels in Hcy in the culture medium inhibit expression of endothelial dimethylarginine dimethylaminohydrolase (DDAH), the main catabolic enzyme of ADMA. This could explain the increase in plasma ADMA observed in experimental hyperhomocysteinemia (Dayal et al. 2008; Stuhlinger et al. 2001). Interestingly, in humans, an increase in plasma ADMA has been reported in acute hyperhomocysteinemia, but healthy subjects with isolated hyperhomocysteinemia have similar levels of plasma ADMA compared to matched controls (Antoniades et al. 2006). Since methionine in the form of S-adenosylmethionine (SAM) provides methyl groups in transmethylation processes, including those concerning protein amino acids residues, it has been hypothesized that only acute hyperhomocysteinemia induced by methionine loading could increase circulating ADMA concentrations (Antoniades et al. 2006). In contrast, a study in healthy mice showed that hyperhomocysteinemia induced by chronic methionine intake for 10 weeks was associated with an impairment of vascular function and a decrease in hepatic DDAH expression, yet with no changes in plasma levels of ADMA (Dayal et al. 2008). Similarly, several studies in humans and animals models did not observe any change in plasma ADMA concentration, even in endothelial dysfunction induced by acute hyperhomocysteinemia (Doshi et al. 2005; Mariotti et al. 2006; Wanby et al. 2003; Tousoulis et al. 2011).
Despite the increasing interest in the DDAH/ADMA pathway as a therapeutic target in cardiovascular disease, the role that ADMA could play in the initiation of endothelial dysfunction induced by hyperhomocysteinemia is not completely understood. Therefore, we sought to study ADMA metabolism in a rat model of endothelial dysfunction induced by acute hyperhomocysteinemia by means of a methionine load. Our main focus was on the DDAH enzyme that hydrolyses ADMA to dimethylamine (DMA) and l-citrulline, on ADMA synthesis in proteins and their proteolysis to release free ADMA.
Materials and methods
Animals
Male Wistar-Kyoto rats (9 weeks old; Harlan, France) were housed (four per cage) in a temperature-controlled room (22 ± 2 °C) on a 12:12-h light/dark cycle, with free access to tap water and standard rat chow. A total of 30 rats were used in a series of studies as described below. The study protocol was approved by the Regional (Ile de France Sud) Animal Care and Ethics Committee. All procedures were performed in accordance with the guidelines issued by the French National Animal Care Committee.
Study design
During 3 weeks before the methionine load studies, 16 rats were accustomed to the experimental conditions including handling, gastric gavage, restraining and vascular reactivity testing as described below. The methionine loading was 3 g methionine/kg as solution in water. A 2.5-mL methionine-free drinking water (i.e., the vehicle) was used as a control to ascertain that variations in circadian rhythm and experimental conditions (e.g., due to handling, gavage and blood sampling) do not affect post-loading measurements. All tests began after a 12-h fast. An indwelling catheter, filled with heparinized (50 U/mL) 2 % NaCl, was inserted into a lateral tail vein to allow frequent blood sampling and injection with minimal discomfort for the conscious animal. Venous blood (500 µL) was sampled and vascular reactivity was tested before (0 h), and 2, 4 and 6 h after the methionine load (n = 10) or the vehicle (n = 6). Fresh whole blood was used immediately for determination of blood glucose. All other blood samples were drawn into tubes pre-chilled with sodium EDTA or heparin and centrifuged immediately (5 min, 3500×g, 4 °C). Plasma samples were stored at −80 °C for further determinations as described below. At the end of the experiment (i.e., 6 h), rats were sacrificed, abdominal aorta and liver tissues were collected and immediately stored at −80 °C.
Vascular reactivity test
In a quiet and temperature-controlled room (26 ± 2 °C), systolic and diastolic blood pressures were measured in 16 conscious rats (methionine load n = 10; vehicle n = 6) by volume–pressure recordings with a plethysmographic sensor and an automated tail-cuff system (XBP1000, Kent Scientific), a specific, very accurate, validated method (Feng et al. 2008). After 10 min of adaptation to inflation and deflation of the occlusion cuff in the restrainer, mean arterial pressure (MAP), calculated as one-third systolic plus two-third diastolic blood pressure, was derived from an average of 10 consecutive blood pressure recordings per rat. Vascular reactivity was measured as the transient decrease in MAP response after the i.v. administration of a 20-mg/kg acetylcholine (Ach) bolus, which has been shown to be predominantly NO-mediated, as previously described (Magne et al. 2009). For each pre- (0 h) and post-methionine load (2, 4, and 6 h) determination on each rat, the vascular reactivity was calculated as the mean response to 3 consecutive i.v. administrations of an Ach bolus, with a minimum of 5 min between each injection. Vascular reactivity was expressed as the relative decrease in blood pressure, i.e., as [(MAP measured immediately after Ach injection—MAP measured before Ach injection)/MAP measured before Ach injection] × 100.
Biochemical analysis
Plasma ADMA and SDMA were determined by HPLC after derivatization with ortho-phthaldialdehyde (OPA) reagent as described elsewhere (Teerlink et al. 2002). For total low molecular weight thiol determination, plasma was first treated with triphenylphosphine, deproteinized with sulfosalicylic acid, and total cysteine (Cys), homocysteine (Hcy) and glutathione (GSH) were assayed by HPLC using pre-column derivatization with 4-aminosulfonyl-7-fluoro-2,13-benzoxadiazole as described elsewhere (Santa et al. 2006). The analysis was also conducted without prior reduction with triphenylphosphine for the determination of the reduced form of those thiols.
Quantitative RT-PCR
Total RNA was extracted from abdominal aorta and liver tissues using Trizol reagent (Invitrogen). Four hundred nanograms of total RNA were converted into cDNA using the High Capacity cDNA Reverse-Transcription Kit (Applied Biosystems) on a PTC-200 thermocycler (MJ Research). RT-PCR amplifications were performed with a Prism7300 sequence detection system using SYBRGreen MasterMix (Applied Biosystems). DDAH1, DDAH2, cathepsin D, cathepsin E2 and ubiquitin mRNA levels were expressed as their ratio to ribosomal 18S RNA levels. Ubiquitin, cathepsin D and E2 are key markers of the ubiquitin proteasome system and the lysosomal proteolysis.
Statistical analysis
Data were expressed as mean ± standard error. For the post-challenge studies (with repeated measurements after methionine or vehicle load), data were analysed using mixed-model procedures for repeated measurements under SAS (SAS Institute), with challenge (methionine vs. vehicle) and time (0, 2, 4, 6 h after the challenge) used as fixed effects. Interactions between challenge and time were also tested. When a fixed effect was significant, post hoc testing was performed under the mixed-model with the Tukey–Kramer adjustment. A P value <0.05 was considered statistically significant.
Results
Vascular reactivity and total plasma homocysteine
As expected, methionine but not vehicle load markedly impaired vascular reactivity (time × challenge effect, P < 0.001; Fig. 1), with a 30 % decrease at 4 and 6 h after the methionine load, as compared to the baseline value (Fig. 1). Compared to the baseline values, methionine administration induced a sustained increase in tHCy as of 2 h after gastric gavage from 10.1 ± 0.5 µM at the baseline to 54.9 ± 1.9 µM at 4 h (P < 0.0001; Fig. 2a). Plasma total Cys and total GSH did not significantly change with time, and there were no differences between methionine and vehicle load (Fig. 2b, c). The post-methionine changes in plasma concentrations of the reduced forms of Cys, Hcy and GSH were similar to those of the concentrations of total (reduced plus oxidized) forms (data not shown).

Vascular reactivity before (0 h), 2, 4 and 6 h after methionine load or vehicle (water). Values are mean ± SE (methionine load n = 10; vehicle n = 6), *P < 0.05 vs values after vehicle administration at the same time point

Plasma total homocysteine (a), total cysteine (b), total glutathione (c) before (0 h), 2, 4 and 6 h after methionine load or vehicle (water). Values are mean ± SE (methionine load n = 10; vehicle n = 6). **P < 0.001 vs. vehicle
Circulating ADMA and SDMA, and tissue DDAH
When compared to the values found at the baseline (0 h), plasma ADMA and SDMA transiently decreased 2 h after methionine but not vehicle load (time × challenge effect, P < 0.001; Fig. 3a, b). Six hours after methionine application, hepatic DDAH1 mRNA expression levels were significantly lower than those after the vehicle administration (P < 0.05; Fig. 4a). Aortic DDAH2 mRNA expression did not significantly differ between methionine and vehicle treatment (Fig. 4b).

Plasma ADMA (a) and SDMA (b) before, 2, 4 and 6 h after methionine load or vehicle (water). Values are mean ± SE (methionine load n = 10; vehicle n = 6). *P < 0.05 vs. vehicle

Hepatic DDAH1 (a) and aortic DDAH2 (b) mRNA expression relative to ribosomal 18S RNA levels, 6 h after methionine load or vehicle (water). Values are mean ± SE (methionine load n = 10; vehicle n = 6). *P < 0.05 vs. vehicle
Six hours after the methionine load, hepatic mRNA expression levels of cathepsin D and ubiquitin were significantly lower compared to vehicle application (P < 0.001; Fig. 5a and P < 0.05; Fig. 5b). Hepatic cathepsin E2 mRNA expression did not significantly differ between the two treatment modes (Fig. 5c).

Hepatic cathepsin D (a), cathepsin E2 (b), ubiquitin (c) mRNA expression relative to ribosomal 18S RNA levels, 6 h after methionine load or vehicle (water). Values are mean ± SE (methionine load n = 10; vehicle n = 6). *P < 0.05, **P < 0.001 vs. vehicle
Discussion
The most important result of the present study is that in healthy rats methionine loading, an experimental model of endothelial dysfunction induced by acute experimental hyperhomocysteinemia, resulted in an pronounced decrease in plasma ADMA and SDMA concentrations. This observation was unexpected because methionine is the precursor of the methyl donor SAM, which is the cofactor utilized in the N-guanidine dimethylation of arginine residues in proteins by protein arginine methyltransferase (PRMT) (Teerlink et al. 2009). Because ADMA and SDMA inhibit NO synthesis (Tsikas et al. 2000a), the endothelial dysfunction measured as a decrease in endothelium-related vascular reactivity, cannot be attributed to these arginine derivatives in the present study. However, since there is a complex compartmentalisation in the vessel wall (endothelial cells) of ADMA and NOS, we cannot entirely discard a possible role of ADMA in the initiation of endothelial dysfunction (Lorin et al. 2014). Our observation does not support the notion that a methionine load stimulates expression or activity of PRMT, at least not in an extent that could result in an increase in circulating ADMA (Antoniades et al. 2006). Indeed in case of high methionine intake, it is considered that the activation of glycine N-methyltransferase in the liver results in driving largely the methyl of SAM toward the conversion of glycine to sarcosine, enabling the catabolism of methionine in excess and the disposal of the methyl load (Brosnan et al. 2007). Therefore, it remains uncertain to what extent a methionine load does result in a marked increase in methylation of arginine residues in proteins.
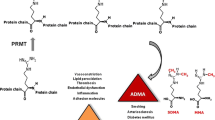
Dimethylation of arginine residues in proteins generates ADMA- and SDMA-containing proteins. It is generally assumed that proteolysis of such proteins releases free ADMA and SDMA which are then distributed throughout the organism and act in different types of cell including endothelial cells (Pope et al. 2009). Methionine administration in our study resulted in the inhibition of the main intracellular proteolytic system, the ubiquitin–proteasome system, a key system involved in proteolysis. Hyperhomocysteinemia can also alter the ubiquitin–proteasome activity in the heart where protein aggregates accumulate (Derouiche et al. 2014). This is also likely to have occurred in the present study. Furthermore, our study reveals a significant down-regulation of hepatic cathepsin D, an aspartic protease involved in lysosomal proteolysis. Cathepsin D is a major component of the lysosome and is actively involved in the maturation and the proteolytic process of this organelle (Zaidi et al. 2008). The ubiquitin–proteasome system and the cathepsin autophagy/lysosomal pathway are considered two main proteolytic degradation systems in eukaryotic cells and were recently shown to contribute to the biosynthesis of free ADMA and SDMA (Shirakawa et al. 2011). Therefore, the parallel decrease in circulating ADMA and SDMA concentration seen in our study upon methionine administration is likely to have resulted from a decreased activity of proteolytic systems.
SDMA is excreted almost unchanged in the urine and is not hydrolysed by DDAH. About 10 % of ADMA are excreted unchanged and the remaining fraction of about 90 % is excreted as dimethylamine (DMA) after hydrolysis of ADMA by DDAH (Siroen et al. 2006). In vitro studies have shown that Hcy can inhibit the expression and the activity of DDAH (Dayal et al. 2008; Dayoub et al. 2003; Stuhlinger et al. 2003). Hcy reacts with cysteine moieties of proteins and enzymes to form mixed disulfides, thus altering their function and expression (Di Simplicio et al. 2005). The inhibitory action of Hcy towards DDAH is considered to be due to the reaction of the SH group of Hcy with the SH group of a cysteine residue in the active center of DDAH. Although we did measure the mRNA and not the protein level or activity, the significant down-regulation of the expression of DDAH1, the predominant enzyme degrading ADMA to DMA and l-citrulline in the liver, and the parallel decrease of circulating ADMA and SDMA concentrations seen in our study argue against an inhibitory action of methionine-derived Hcy in the metabolism of ADMA by DDAH in the liver and most likely in the kidney. Because we did not measure in our study ADMA, DMA and SDMA in the urine, we cannot strictly rule out that a decrease in circulating ADMA and SDMA concentrations may be ascribed in part to their enhanced excretion in the urine, but such a change in the clearance of methyl-arginine is unlikely.
It should be noted that the relation between homocysteine, plasma methyl-arginine, and the likely underlying metabolic determinant as studied here pertain to the experimental model used, i.e., acute hyperhomocysteinemia, and could not be readily extrapolated to situations of chronic hyperhomocysteinemia.
Elevated plasma ADMA levels are associated with an increased risk of incidence for cardiovascular events (Böger 2006). More recently, SDMA, the structural isomer of ADMA, which can also inhibit NO synthesis from l-arginine (Tsikas et al. 2000b), has also been shown to be associated with cardiovascular risk (Bode-Böger et al. 2006; Gore et al. 2013; Schwedhelm et al. 2014). Our study shows that the concentration of these two NOS inhibitors in rat plasma acutely decreases each by about 20 % upon methionine administration. A definite explanation for this observation cannot be given by our study. Impaired proteolysis of ADMA- and SDMA-containing proteins paired with unaffected hepatic and renal metabolism and elimination of free ADMA and SDMA may be the most likely explanation of our findings. Delineation of underlying mechanisms would require measurement of additional biochemical parameters including ADMA- and SDMA-containing proteins in body fluids and tissues, which have not been measured in the present study because of the unavailability of suitable analytical methods. Nevertheless, our study suggests that acutely homocysteine-induced endothelial dysfunction in rats is most likely not associated with ADMA and SDMA. The mechanisms underlying the association of ADMA and SDMA with cardiovascular risk are still unresolved.
Abbreviations
- Ach:
-
Acetylcholine
- ADMA:
-
Asymmetric dimethylarginine
- DDAH:
-
Dimethylarginine dimethylaminohydrolase
- DMA:
-
Dimethylarginine
- MAP:
-
Mean arterial pressure
- NO:
-
Nitric oxide
- PRMT:
-
Protein arginine methyltransferase
- ROS:
-
Reactive oxygen species
- SAM:
-
S-adenosylmethionine
- SDMA:
-
Symmetric dimethylarginine
- tHCy:
-
Total plasma homocysteine
References
Andreotti F, Burzotta F, Manzoli A, Robinson K (2000) Homocysteine and risk of cardiovascular disease. J Thromb Thrombolysis 9(1):13–21
Antoniades C, Tousoulis D, Marinou K, Vasiliadou C, Tentolouris C, Bouras G, Pitsavos C, Stefanadis C (2006) Asymmetrical dimethylarginine regulates endothelial function in methionine-induced but not in chronic homocystinemia in humans: effect of oxidative stress and proinflammatory cytokines. Am J Clin Nutr 84(4):781–788
Bellamy MF, McDowell IF, Ramsey MW, Brownlee M, Bones C, Newcombe RG, Lewis MJ (1998) Hyperhomocysteinemia after an oral methionine load acutely impairs endothelial function in healthy adults. Circulation 98(18):1848–1852
Bode-Böger SM, Scalera F, Kielstein JT, Martens-Lobenhoffer J, Breithardt G, Fobker M, Reinecke H (2006) Symmetrical dimethylarginine: a new combined parameter for renal function and extent of coronary artery disease. J Am Soc Nephrol 17(4):1128–1134. doi:10.1681/asn.2005101119
Böger RH (2006) Asymmetric dimethylarginine (ADMA): a novel risk marker in cardiovascular medicine and beyond. Ann Med 38(2):126–136. doi:10.1080/07853890500472151
Böger RH, Bode-Böger SM, Sydow K, Heistad DD, Lentz SR (2000) Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in monkeys with hyperhomocyst(e)inemia or hypercholesterolemia. Arterioscler Thromb Vasc Biol 20(6):1557–1564
Böger RH, Lentz SR, Bode-Böger SM, Knapp HR, Haynes WG (2001) Elevation of asymmetrical dimethylarginine may mediate endothelial dysfunction during experimental hyperhomocyst(e)inaemia in humans. Clin Sci (Lond) 100(2):161–167
Brosnan JT, da Silva R, Brosnan ME (2007) Amino acids and the regulation of methyl balance in humans. Curr Opin Clin Nutr Metab Care 10(1):52–57. doi:10.1097/MCO.0b013e3280110171
Casas JP, Bautista LE, Smeeth L, Sharma P, Hingorani AD (2005) Homocysteine and stroke: evidence on a causal link from mendelian randomisation. Lancet 365(9455):224–232. doi:10.1016/s0140-6736(05)17742-3
Chambers JC, Obeid OA, Kooner JS (1999) Physiological increments in plasma homocysteine induce vascular endothelial dysfunction in normal human subjects. Arterioscler Thromb Vasc Biol 19(12):2922–2927
Dayal S, Lentz SR (2008) Murine models of hyperhomocysteinemia and their vascular phenotypes. Arterioscler Thromb Vasc Biol 28(9):1596–1605. doi:10.1161/atvbaha.108.166421
Dayal S, Rodionov RN, Arning E, Bottiglieri T, Kimoto M, Murry DJ, Cooke JP, Faraci FM, Lentz SR (2008) Tissue-specific downregulation of dimethylarginine dimethylaminohydrolase in hyperhomocysteinemia. Am J Physiol Heart Circ Physiol 295(2):H816–H825. doi:10.1152/ajpheart.01348.2007
Dayoub H, Achan V, Adimoolam S, Jacobi J, Stuehlinger MC, Wang BY, Tsao PS, Kimoto M, Vallance P, Patterson AJ, Cooke JP (2003) Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation 108(24):3042–3047. doi:10.1161/01.cir.0000101924.04515.2e
De Bree A, Verschuren WM, Kromhout D, Kluijtmans LA, Blom HJ (2002) Homocysteine determinants and the evidence to what extent homocysteine determines the risk of coronary heart disease. Pharmacol Rev 54(4):599–618
den Heijer M, Willems HP, Blom HJ, Gerrits WB, Cattaneo M, Eichinger S, Rosendaal FR, Bos GM (2007) Homocysteine lowering by B vitamins and the secondary prevention of deep vein thrombosis and pulmonary embolism: a randomized, placebo-controlled, double-blind trial. Blood 109(1):139–144. doi:10.1182/blood-2006-04-014654
Derouiche F, Bole-Feysot C, Naimi D, Coeffier M (2014) Hyperhomocysteinemia-induced oxidative stress differentially alters proteasome composition and activities in heart and aorta. Biochem Biophys Res Commun 452(3):740–745. doi:10.1016/j.bbrc.2014.08.141
Di Simplicio P, Frosali S, Priora R, Summa D, Cherubini Di Simplicio F, Di Giuseppe D, Di Stefano A (2005) Biochemical and biological aspects of protein thiolation in cells and plasma. Antioxid Redox Signal 7(7–8):951–963. doi:10.1089/ars.2005.7.951
Doshi S, McDowell I, Goodfellow J, Stabler S, Böger R, Allen R, Newcombe R, Lewis M, Moat S (2005) Relationship between S-adenosylmethionine, S-adenosylhomocysteine, asymmetric dimethylarginine, and endothelial function in healthy human subjects during experimental hyper- and hypohomocysteinemia. Metabolism 54(3):351–360. doi:10.1016/j.metabol.2004.09.015
Feng M, Whitesall S, Zhang Y, Beibel M, D’Alecy L, DiPetrillo K (2008) Validation of volume-pressure recording tail-cuff blood pressure measurements. Am J Hypertens 21(12):1288–1291. doi:10.1038/ajh.2008.301
Fu YF, Xiong Y, Guo Z (2005) A reduction of endogenous asymmetric dimethylarginine contributes to the effect of captopril on endothelial dysfunction induced by homocysteine in rats. Eur J Pharmacol 508(1–3):167–175. doi:10.1016/j.ejphar.2004.11.063
Gore MO, Luneburg N, Schwedhelm E, Ayers CR, Anderssohn M, Khera A, Atzler D, de Lemos JA, Grant PJ, McGuire DK, Böger RH (2013) Symmetrical dimethylarginine predicts mortality in the general population: observations from the Dallas heart study. Arterioscler Thromb Vasc Biol 33(11):2682–2688. doi:10.1161/atvbaha.113.301219
Hanratty CG, McGrath LT, McAuley DF, Young IS, Johnston GD (2001) The effects of oral methionine and homocysteine on endothelial function. Heart 85(3):326–330
Jakubowski H (2006) Pathophysiological consequences of homocysteine excess. J Nutr 136(6 Suppl):1741S–1749S
Jamison RL, Hartigan P, Kaufman JS, Goldfarb DS, Warren SR, Guarino PD, Gaziano JM, Veterans Affairs Site I (2007) Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: a randomized controlled trial. JAMA 298(10):1163–1170. doi:10.1001/jama.298.10.1163
Lentz SR, Haynes WG (2004) Homocysteine: is it a clinically important cardiovascular risk factor? Clevel Clin J Med 71(9):729–734
Lorin J, Zeller M, Guilland JC, Cottin Y, Vergely C, Rochette L (2014) Arginine and nitric oxide synthase: regulatory mechanisms and cardiovascular aspects. Mol Nutr Food Res 58(1):101–116. doi:10.1002/mnfr.201300033
Loscalzo J (2006) Homocysteine trials–clear outcomes for complex reasons. N Engl J Med 354(15):1629–1632. doi:10.1056/NEJMe068060
Magne J, Huneau JF, Delemasure S, Rochette L, Tome D, Mariotti F (2009) Whole-body basal nitric oxide production is impaired in postprandial endothelial dysfunction in healthy rats. Nitric Oxide 21(1):37–43. doi:10.1016/j.niox.2009.04.003
Mariotti F, Hammiche A, Blouet C, Dare S, Tome D, Huneau JF (2006) Medium-term methionine supplementation increases plasma homocysteine but not ADMA and improves blood pressure control in rats fed a diet rich in protein and adequate in folate and choline. Eur J Nutr 45(7):383–390. doi:10.1007/s00394-006-0611-1
Pope AJ, Karuppiah K, Cardounel AJ (2009) Role of the PRMT-DDAH-ADMA axis in the regulation of endothelial nitric oxide production. Pharmacol Res 60(6):461–465. doi:10.1016/j.phrs.2009.07.016
Santa T, Aoyama C, Fukushima T, Imai K, Funatsu T (2006) Suppression of thiol exchange reaction in the determination of reduced-form thiols by high-performance liquid chromatography with fluorescence detection after derivatization with fluorogenic benzofurazan reagent, 7-fluoro-2,1,3-benzoxadiazole-4-sulfonate and 4-aminosulfonyl-7-fluoro-2,1,3-benzoxadiazole. Biomed Chromatogr 20(6–7):656–661. doi:10.1002/bmc.683
Schwedhelm E, Wallaschofski H, Atzler D, Dorr M, Nauck M, Volker U, Kroemer HK, Volzke H, Böger RH, Friedrich N (2014) Incidence of all-cause and cardiovascular mortality predicted by symmetric dimethylarginine in the population-based study of health in Pomerania. PLoS One 9(5):e96875. doi:10.1371/journal.pone.0096875
Shirakawa T, Kako K, Shimada T, Nagashima Y, Nakamura A, Ishida J, Fukamizu A (2011) Production of free methylarginines via the proteasome and autophagy pathways in cultured cells. Mol Med Rep 4(4):615–620. doi:10.3892/mmr.2011.488
Siroen MP, Teerlink T, Nijveldt RJ, Prins HA, Richir MC, van Leeuwen PA (2006) The clinical significance of asymmetric dimethylarginine. Annu Rev Nutr 26:203–228. doi:10.1146/annurev.nutr.26.061505.111320
Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP (2001) Homocysteine impairs the nitric oxide synthase pathway: role of asymmetric dimethylarginine. Circulation 104(21):2569–2575
Stuhlinger MC, Oka RK, Graf EE, Schmolzer I, Upson BM, Kapoor O, Szuba A, Malinow MR, Wascher TC, Pachinger O, Cooke JP (2003) Endothelial dysfunction induced by hyperhomocyst(e)inemia: role of asymmetric dimethylarginine. Circulation 108(8):933–938
Teerlink T, Nijveldt RJ, de Jong S, van Leeuwen PA (2002) Determination of arginine, asymmetric dimethylarginine, and symmetric dimethylarginine in human plasma and other biological samples by high-performance liquid chromatography. Anal Biochem 303(2):131–137. doi:10.1006/abio.2001.5575
Teerlink T, Luo Z, Palm F, Wilcox CS (2009) Cellular ADMA: regulation and action. Pharmacol Res 60(6):448–460. doi:10.1016/j.phrs.2009.08.002
Tousoulis D, Bouras G, Antoniades C, Marinou K, Papageorgiou N, Miliou A, Hatzis G, Stefanadi E, Tsioufis C, Stefanadis C (2011) Methionine-induced homocysteinemia impairs endothelial function in hypertensives: the role of asymmetrical dimethylarginine and antioxidant vitamins. Am J Hypertens 24(8):936–942. doi:10.1038/ajh.2011.65
Tsikas D, Böger RH, Sandmann J, Bode-Böger SM, Frölich JC (2000a) Endogenous nitric oxide synthase inhibitors are responsible for the l-arginine paradox. FEBS Lett 478(1–2):1–3
Tsikas D, Sandmann J, Savva A, Luessen P, Böger RH, Gutzki FM, Mayer B, Frölich JC (2000b) Assessment of nitric oxide synthase activity in vitro and in vivo by gas chromatography-mass spectrometry. J Chromatogr B Biomed Sci Appl 742(1):143–153
Wanby P, Brattstrom L, Brudin L, Hultberg B, Teerlink T (2003) Asymmetric dimethylarginine and total homocysteine in plasma after oral methionine loading. Scand J Clin Lab Invest 63(5):347–353
Zaidi N, Maurer A, Nieke S, Kalbacher H (2008) Cathepsin D: a cellular roadmap. Biochem Biophys Res Commun 376(1):5–9. doi:10.1016/j.bbrc.2008.08.099
Zylberstein DE, Bengtsson C, Bjorkelund C, Landaas S, Sundh V, Thelle D, Lissner L (2004) Serum homocysteine in relation to mortality and morbidity from coronary heart disease: a 24-year follow-up of the population study of women in Gothenburg. Circulation 109(5):601–606. doi:10.1161/01.cir.0000112581.96154.ea
Acknowledgments
We thank Maëlle Robert for her contribution to the experimentation and Dominique Hermier for the help with the collection of the tissue samples. This work was supported by the French Ministry of Research. J. Magné is supported by the Swedish Heart–Lung Foundation, the Fredrik and Ingrid Thuring Foundation, and the Lars Hiertas Minne Foundation.
Conflict of interest
None of the authors had a conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the guidelines issued by the French National Animal Care Committee at which the studies were conducted.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Magné, J., Huneau, JF., Borderie, D. et al. Plasma asymmetric and symmetric dimethylarginine in a rat model of endothelial dysfunction induced by acute hyperhomocysteinemia. Amino Acids 47, 1975–1982 (2015). https://doi.org/10.1007/s00726-015-1959-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-015-1959-4