
Abstract
Here we review the strategies for the solid-phase synthesis of peptides starting from the side chain of the C-terminal amino acid. Furthermore, we provide experimental data to support that C-terminal and side-chain syntheses give similar results in terms of purity. However, the stability of the two bonds that anchor the peptide to the polymer may determine the overall yield and this should be considered for the large-scale production of peptides. In addition, resins/linkers which do not subject to side reactions can be preferred for some peptides.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The importance of peptides in modern science has grown exponentially in recent years. In addition to that, peptides are considered a firm alternative to small molecules for the treatment of a large number of human and animal diseases (Albericio and Kruger 2012; Kaspar and Reichert 2013; Scognamiglio et al. 2013; Gongora-Benitez et al. 2014). The cosmetic and nutraceutical industries are also introducing peptides into their products (Mentel et al. 2012; Udenigwe and Howard 2013). Furthermore, peptides are also used in the development of drug delivery systems and diagnostic kits (Li et al. 2012; Vasconcelos et al. 2013). Finally, new biomaterials for a broad range of applications are currently prepared from peptides (Nune et al. 2013; Menzel 2013). The introduction of these biomolecules into our everyday lives (Zompra et al. 2009; Chandrudu et al. 2013) has unquestionably been fuelled by the development and optimization of the solid-phase synthetic strategy, first described by Merrifield (Merrifield 1963).
In brief, the solid-phase peptide synthesis (SPPS) is based on the concourse of a supported protecting group—a polymeric support—that facilitates the stepwise elongation of this biopolymer through sequential steps of coupling and deprotection of protected amino acids, thus allowing the use of large excess of reagents. At the end of the synthetic process, a chemical treatment is usually applied to remove the protecting groups and detach the peptide from the resin (Albericio et al. 2011; Gongora-Benitez et al. 2013).
All peptides have the following four types of functional groups which can in principle be used for attachment to the polymeric support: (1) C-terminal function (C to N strategy); (2) N-terminal function (N to C strategy); (3) backbone; and (4) side chain (if a trifunctional amino acid is present) (Fig. 1).

Schematic representation of a peptide showing the anchoring sites to the polymeric support
SPPS through the C-terminal function is the most predominant strategy for the preparation of peptides because the formation of the peptide bond usually requires the activation of the carboxylic acid component. This is the key component of the reaction, which is the one added in solution and can be used in large excess driving the reaction to completion (Lloyd-Williams et al. 1997; Kates and Albericio 2000). Furthermore, the amino function of the activated amino acid component is commonly protected as a carbamate, whose slight electron-withdrawing effect does not facilitate oxazolone formation—the cause of racemization and poor yields—and does not overly enhance the acidity of the α-proton, thus minimizing the racemization through the enol mechanism (El-Faham and Albericio 2011).
Several attempts of SPPS through N-anchoring have been made (Thieriet et al. 2000). However, the need to activate acid terminal in this strategy jeopardizes its broad use, because potential racemization through oxazolone formation can occur at each step [route (a) in Fig. 2]. Furthermore, diketopiperazine can also form in each step [route (b) in Fig. 2].

Main side reactions during the N to C elongation
Backbone anchoring is normally used when manipulation of the C-terminal function is required and the C-terminal amino acid is not trifunctional (Jensen et al. 1998, 1999). For trifunctional amino acids, except for Arg, side-chain anchoring is preferred due the simplicity of the strategy. The backbone amide linker (BAL) is very useful for the synthesis of C-terminal-modified peptides, such as peptide aldehyde (Kappel and Barany 2005; Boas et al. 2009).
SPPS through side-chain anchoring is used for the following cases:
-
1.
Synthetic comfort It is now widely accepted that the synthesis of peptide amides is more convenient than for their counterpart acids. The formation of the initial amide bond (side chain of Gln and Asn) is generally achieved with better yields and less racemization when compared with ester formation in Wang-type resins. Furthermore, the amide bond is on the whole more stable than the ester bond and, therefore, peptide amides are synthesized with better yields (Albericio et al. 1990; Breipohl et al. 1990).
-
2.
Minimization of side reaction formation The synthesis of C-terminal Cys-containing peptide acids is accompanied by high level of Cys racemization and N-piperidyl-Ala through the formation of didehydro-Ala residue followed by a piperidine Michael addition. In this regard, Barany and co-workers demonstrated that the side anchoring of Fmoc-Cys-OtBu to the solid support through a xanthenyl handle allows minimization of the above-mentioned side reactions in peptide synthesis (Barany et al. 2003; Han and Barany 1997).
Alkoxybenzyl resins and linkers, such as Wang-type, PAL, Rink, BAL, are cleaved by different points during the TFA treatment, thus leading to several carbocations, which can be reattached to the peptide with the consequent formation of side products (Yraola et al. 2004; Cironi et al. 2004). These resins are used for the preparation of both acid and amide peptides. In this regard, the use of chloro-trityl chloride (CTC), developed by Barlos and co-workers, does not show this abnormal cleavage and therefore the synthesis, for instance, of Ser/Thr-NH2 C-terminal peptides through the side-chain anchoring of Fmoc-Ser/Thr-NH2 to CTC resins is advantageous for this kind of peptide (Ziovas et al. 2012; Barlos 2013a, b).
-
3.
On-resin cyclization Cyclic peptides can be prepared on solid-phase by the side- chain anchoring while the C-terminal acid bears an orthogonal protecting group. The protecting group is then removed after the elongation of the sequence to be further activated for rendering the macrolactamization with a free amino function (N-terminal or side-chain amino function) (Kates et al. 1994; Rovero 2000).
-
4.
Manipulation of the C-terminal function A similar strategy to that used for on- resin cyclization can be used to prepare modified peptides such as the C-terminal thioesters required for chemical ligation, after reaction of the activated C-terminal carboxylic acid with the corresponding thiol (Diaz-Rodriguez et al. 2012; Ajish et al. 2009; Ficht et al. 2008; Tulla-Puche et al. 2004; Alsin et al. 2000). In the case of other modified peptides, such as the C-terminal p-nitroanilide, where the precursor is a poor nucleophile, the synthesis starts at the residue n-1 and at the end the C-terminal amino acid is incorporated in form of the p-nitroanilide, which is prepared in solution after cleavage of the peptide from the resin.
The following table describes the resins and/or linkers used for side-chain anchoring, with representative references (Table 1).
In addition to the four cases described above regarding the use of side-chain vs. C-terminal SPPS, there is the question as to whether side-chain anchoring has some additional (dis)advantages over the C-terminal strategy with respect to the peptide skeleton arrangement in relation to the polymer. Thus, in the 90 s, Larsen and Holm described a new concept, sequence-assisted peptide synthesis (SAPS) (Larsen and Holm 1996). This approach is based on the introduction of certain C-terminus-positioned short sequences (Lys)n, which induce a structure in a subsequent peptide chain that can lead to improved synthesis of difficult sequences. This concept was demonstrated for the synthesis of several (Ala)n and (Thr-Val)n peptides, as well as other natural sequences corresponding to acyl carrier protein and insulin (Larsen and Holm 1998a). This concept was further reinforced when they showed that the use of 4-methoxymandelic acid as a Wang-type handle, which binds the polymer to the peptide through a three-atom moiety, renders better results than when a typical Wang linker, such as the 4-hydroxymethylphenoxypropionic acid, is used (Larsen and Holm 1998b). If the SAPS concept is correct, side-chain anchoring could impair bonding between the polymer and the peptide chain.
Although we demonstrated that the synthesis of Thymosin α1 via side-chain anchoring of the C-terminal Asn/Asp through the β-carboxylic acid of Asp to a Rink resin performs better than through the α-carboxylic acid of Asn to a CTC resin, this result does not validate the hypothesis that side-chain anchoring is more favorable than that of C-terminal synthesis, because more than one parameter was changed (linker and resin) (Garcia-Ramos et al. 2009).
Bearing all these factors in mind, we designed a simple experiment to determine whether there are any differences between C-terminal and side-chain anchoring. For this purpose, we synthesized a Glu C-terminal peptide. Glu or Asp is the most neutral amino acids for this experiment, because the same resin and the same protection for the remaining carboxylic acid can be used for both syntheses. As a model, the 15 amino acid peptide HIV-1 Rev (91–105) was chosen (Fig. 3).

Sequence of HIV-1 Rev (91–105)
Syntheses were performed by applying methods extensively used in our laboratories (Pelay-Gimeno et al. 2013). For this purpose, we anchored Fmoc-Glu(OtBu)-OH and Fmoc-Glu-OtBu (0.6 equiv.) to CTC resin (1.6 mmol/g) to render the starting resins [Fmoc-Glu(OtBu)-O-CTC-resin, 202 mg, 0.77 mmol/g and Fmoc-Glu(O-CTC-resin)-OtBu, 204 mg, 0.79 mmol/g]. Elongation of the peptide chain was carried out consecutively with Fmoc-Val-OH (3 equiv.), Fmoc-Leu-OH (3 equiv.), Fmoc-Ile-OH (3 equiv.), Fmoc-Gln(Trt)-OH (3 equiv.), Fmoc-Pro-OH (3 equiv.), Fmoc-Ser(tBu)-OH (3 equiv.), Fmoc-Gly-OH (3 equiv.), Fmoc-Val-OH (3 equiv.), Fmoc-Gly-OH (3 equiv.), Fmoc-Gln(Trt)-OH (3 equiv.), Fmoc-Thr(tBu)-OH (3 equiv.), Fmoc-Gly-OH (3 equiv.), Fmoc-Ser(tBu)-OH (3 equiv.), Fmoc-Thr(tBu)-OH (3 equiv.), using COMU (3 equiv.), OxymaPure (3 equiv.) and DIEA (6 equiv.) for 1 h in DMF. No recouplings were done. At the end of the syntheses, the global deprotection and cleavage of the peptide were carried out with TFA–H2O–TIS (95:2.5:25) for 1 h. After precipitation of the peptide in cold ether and further washing with ether, it was dissolved in AcOH–H2O (9:1) and lyophilized to give 180 mg (76 % overall yield) for C-terminal anchoring and 206 mg (87 % yield) for side-chain anchoring.
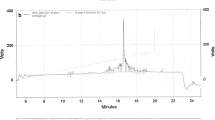
HPLC revealed that the crude peptides showed (Fig. 4) virtually identical purity (97.3 % for C-terminal anchoring and 97.5 % for side-chain anchoring), the main impurity being the same for both syntheses. Furthermore, the EI-MS showed a similar profile for both peptides.

HPLC and HPLC–MS of HIV-1 Rev (91–105); a from C-terminal anchoring; b from side-chain anchoring
We conclude that, in terms of purity, there are no major advantages of performing peptide synthesis through C-terminal vs./and side-chain anchoring. This conclusion is supported by the purity of the final products achieved. However, the better yield obtained (by approx. 10 %), which can be attributed to the better stability of the ester bond anchoring through the β-carboxylic (less acid and therefore worse leaving group) vs. the α-acid group, makes side-chain anchoring highly suitable for large-scale peptide synthesis. This improved stability can be translated into the preparation of longer peptides. In addition, this strategy is very convenient for the synthesis of cyclic and C-terminal-modified peptides. Finally, side-chain anchoring could allow the use of resins/linkers that do not subject to side reactions, which will make this strategy preferred in front of the C-terminal anchoring.
References
Ajish KKS, Harpaz Z, Haj-Yahya M, Brik A (2009) Side-chain assisted ligation in protein synthesis. Bioorg Med Chem Lett 19:3870–3874
Albericio F, Kruger HG (2012) Therapeutic peptides. Future Med Chem 4:1527–1531
Albericio F, van Abel R, Barany G (1990) Solid-phase synthesis of peptides with C-terminal asparagine or glutamine. Int J Pept Protein Res 35:284–286
Albericio F, Tulla-Puche J, Kates SA (2011) Fmoc methodology: cleavage from the resin and final deprotection. In: Hughes AB (ed) Amino acids, peptides and proteins in organic chemistry. Building blocks, catalysis and coupling chemistry, vol 3. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, pp 349–369
Alcaro MC, Orfei M, Chelli M, Ginanneschi M, Papini AM (2003) Solid-phase approach to the synthesis of cyclen scaffolds from cyclotetrapeptides. Tetrahedron Lett 44:5217–5219
Alsin J, Yokum SS, Albericio F, Barany G (2000) A modified backbone amide linker (BAL) solid-phase peptide synthesis strategy accommodating prolyl, N-alkylamino acyl, or histidyl derivatives at the C-terminus. Tetrahedron Lett 41:7277–7280
Alsina J, Rabanal F, Giralt E, Albericio F (1994) Solid-phase synthesis of “head-to-tail” cyclic peptides via lysine side-chain anchoring. Tetrahedron Lett 35:9633–9636
Alsina J, Chiva C, Rabanal F, Giralt E, Albericio F (1997) Active carbonate resins for solid-phase synthesis through the anchoring of an hydroxyl function. Tetrahedron Lett 38:883–886
Alsina J, Rabanal F, Chiva C, Giralt E, Albericio F (1998) Active carbonate resins: application to the solid-phase synthesis of alcohol, carbamate, and cyclic peptides. Tetrahedron 54:10125–10152
Barany G, Han Y, Hargittai B, Liu R-Q, Varkey JT (2003) Side-chain anchoring strategy for solid-phase synthesis of peptide acids with C-terminal cysteine. Biopolymers 71:652–666
Barlos K (2013a) Solid phase peptide synthesis via side chain attachment. PCT Int Appl WO 2013098802 A2 20130704
Barlos K (2013b) Solid phase peptide synthesis of insulin derivatives and analogs using side chain anchored lysine. PCT Int Appl WO 2013156977 A1 20131024
Bernhardt A, Drewello M, Schutkowski M (1997) The solid-phase synthesis of side-chain-phosphorylated peptide-4-nitroanilides. J Pept Res 50:143–152
Beythien J, Barthelemy S, Schneeberger P, White P (2006) A novel solid-phase linker strategy for the side-chain anchoring of arginine: an expeditious route to arginine 7-amido-4-methylcoumarins. Tetrahedron Lett 47:3009–3012
Boas U, Brask J, Jensen KJ (2009) Backbone amide linker in solid-phase synthesis. Chem Rev 109:2092–2118
Breipohl G, Knolle J, Stüber W (1990) Facile SPS of peptides having C-terminal Asn and Gln. Int J Pept Protein Res 35:281–283
Cabrele C, Langer M, Beck-Sickinger AG (1999) Amino acid side chain attachment approach and its application to the synthesis of tyrosine-containing cyclic peptides. J Org Chem 64:4353–4361
Chandrudu S, Simerska P, Toth I (2013) Chemical methods for peptide and protein production. Molecules 18:4373–4388
Cironi P, Tulla-Puche J, Barany G, Albericio F, Alvarez M (2004) Solid-phase syntheses of furopyridine and furoquinoline systems. Org Lett 6:1405–1408
Diaz-Rodriguez V, Mullen DG, Ganusova E, Becker JM, Distefano MD (2012) Synthesis of peptides containing C-terminal methyl esters using trityl side-chain anchoring: application to the synthesis of a-factor and a-factor analogs. Org Lett 14:5648–5651
Edwards PD, Mauger RC, Cottrell KM, Morris FX, Pine KK, Sylvester MA, Scott CW, Furlong ST (2000) Synthesis and enzymatic evaluation of a P1 arginine aminocoumarin substrate library for trypsin-like serine proteases. Bioorg Med Chem Lett 10:2291–2294
El-Faham A, Albericio F (2011) Peptide coupling reagents, more than a letter soup. Chem Rev 111:6557–6602
Ficht S, Payne RJ, Guy RT, Wong C-H (2008) Solid-phase synthesis of peptide and glycopeptide thioesters through side-chain-anchoring strategies. Chem Eur J 14:3620–3629
Garcia O, Nicolas E, Albericio F (2003) Solid-phase synthesis: a linker for side-chain anchoring of arginine. Tetrahedron Lett 44:5319–5321
Garcia-Ramos Y, Giraud M, Tulla-Puche J, Albericio F (2009) Optimized Fmoc solid-phase synthesis of thymosin α1 by side-chain anchoring onto a PEG Resin. Biopolymers (Pept Sci) 92:565–572
Giraud M, Albericio F, Quattrini F, Werbitzky O, Senn K, Williner M (2008) Method for peptide synthesis. PCT Int Appl WO 2008040536 A1 20080410
Gongora-Benitez M, Tulla-Puche J, Albericio F (2013) Handles for Fmoc solid-phase synthesis of protected peptides. ACS Comb Sci 15:217–228
Gongora-Benitez M, Tulla-Puche J, Albericio F (2014) The multifaceted roles of disulfide bonds. Peptides as therapeutics. Chem Rev. doi:10.1021/cr400031z
Graham KAN, Wang Q, Eisenhut M, Haberkorn U, Mier W (2002) A general method for functionalising both the C- and N-terminals of Tyr3-octreotate. Tetrahedron Lett 43:5021–5024
Guillier F, Orain D, Bradley M (2000) Linkers and cleavage strategies in solid-phase organic synthesis and combinatorial chemistry. Chem Rev 100:2091–2157
Han Y, Barany G (1997) Novel S-xanthenyl protecting groups for cysteine and their applications for the N α-9-fluorenylmethyloxycarbonyl (Fmoc) strategy of peptide synthesis. J Org Chem 62:3841–3848
Isied SS, Kuehn CG, Lyon JM, Merrifield RB (1982) Specific peptide sequences for metal ion coordination: solid-phase synthesis of cyclo-(Gly-His)3. J Am Chem Soc 104:2632–2634
Jensen KJ, Alsina J, Songster MF, Vágner J, Albericio F, Barany G (1998) Backbone amide linker (BAL) strategy for solid-phase synthesis of C-terminal modified and cyclic peptides. J Am Chem Soc 120:5441–5452
Jensen KJ, Alsina J, Albericio F, Barany G (1999) Solid-phase synthesis with backbone amide linker (BAL). Chem Eur J 5:2787–2795
Kappel JC, Barany G (2004) Methionine anchoring applied to the solid-phase synthesis of lysine-containing ‘head-to-tail’ cyclic peptides. Lett Pept Sci 10:119–125
Kappel JC, Barany G (2005) Backbone amide linker (BAL) strategy for N α-9-fluorenylmethoxycarbonyl (Fmoc) solid-phase synthesis of peptide aldehydes. J Pept Sci 11:525–535
Kaspar AA, Reichert JM (2013) Future directions for peptide therapeutics. Drug Disc Today 18:807–817
Kaspari A, Schierhorn A, Schutkowski M (1996) Solid-phase synthesis of peptide-4-nitroanilides. Int J Pept Protein Res 48:486–494
Kates SA, Albericio F (eds) (2000) Solid-phase synthesis. A practical guide. Marcel Dekker, New York
Kates SA, Sole NA, Johnson CR, Hudson D, Barany G, Albericio F (1993) A novel, convenient, three-dimensional orthogonal strategy for solid-phase synthesis of cyclic peptides. Tetrahedron Lett 34:1549–1552
Kates SA, Sole NA, Albericio F, Barany G (1994) Solid-phase synthesis of cyclic peptides. In: Anantharamaiah GM (ed) Peptides: design, synthesis, and biological activity (Basava, C. Birkhauser, Boston, pp 39–58
Larsen BD, Holm A (1996) Sequence assisted peptide synthesis (SAPS) (1998) In: Ramage R, Epton R (eds) Peptides 1996, proceedings of the European peptide symposium, 24th, pp 567–568
Larsen BD, Holm A (1998a) Sequence-assisted peptide synthesis (SAPS). J Pept Res 52:470–476
Larsen BD, Holm A (1998b) Improved solid-phase peptide synthesis and agent for use in such synthesis. PCT Int Appl WO 9811125 A1 19980319
Lee Y, Silverman RB (1999) Efficient solid-phase synthesis of compounds containing phenylalanine and its derivatives via side-chain attachment to the polymer support. J Am Chem Soc 121:8407–8408
Li T, He X, Wang Z (2012) The application of peptide functionalized gold nanoparticles. ACS symposium series 1113. Functional nanoparticles for bioanalysis, nanomedicine, and bioelectronic devices, vol 2, pp 55–68
Liu S, Gu W, Lo D, Ding X-Z, Ujiki M, Adrian TE, Soff GA, Silverman RB (2005) N-Methylsansalvamide A peptide analogues. Potent new antitumor agents. J Med Chem 48:3630–3638
Lloyd-Williams P, Albericio F, Giralt E (1997) Chemical approaches to the synthesis of peptides and proteins. CRC, Boca Raton
Masurier N, Zajdel P, Verdie P, Pawlowski M, Amblard M, Martinez J, Subra G (2012) A new highly versatile handle for chemistry on a solid support: the pipecolic linker. Chem Eur J 18:11536–11540
McMurray JS (1991) Solid phase synthesis of a cyclic peptide using Fmoc chemistry. Tetrahedron Lett 32:7679–7682
Mentel M, Schild J, Maczkiewitz U, Koehler T, Farwick M (2012) Innovative peptide technologies for even, young and healthy looking skin. SOFW J 138:22 (24–30, 32–33)
Menzel H (2013) Polypeptide-polymer conjugates. Adv Polym Sci 253:1–36 (bio-synthetic polymer conjugates)
Merrifield RB (1963) Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J Am Chem Soc 85:2149–2154
Nakamura K, Ishii A, Ito Y, Nakahara Y (1999) A novel silyl linker: motif for side-chain tethered approach to solid-phase glycopeptide synthesis. Tetrahedron 55:11253–11266
Nune M, Kumaraswamy P, Krishnan UM, Sethuraman S (2013) Self-assembling peptide nanofibrous scaffolds for tissue engineering: novel approaches and strategies for effective functional regeneration. Curr Protein Pept Sci 14:70–84
Pelay-Gimeno M, Garcia-Martin Y, Martin MJ, Spengler J, Molina-Guijarro JM, Munt S, Francesch AM, Cuevas C, Tulla-Puche J, Albericio F (2013) Total synthesis of pipecolidepsin A, the first member of the homophymine family. Nat Commun 4:2352. doi:10.1038/ncomms3352
Preciado S, Mendive-Tapia L, Torres-Garcia C, Zamudio-Vazquez R, Soto-Cerrato V, Perez-Tomas R, Albericio F, Nicolas E, Lavilla R (2013) Synthesis and biological evaluation of a post-synthetically modified Trp-based diketopiperazine. MedChemComm 4:1171–1174
Rizzi L, Cendic K, Vaiana N, Romeo S (2011) Alcohols immobilization onto 2-chlorotrityl chloride resin under microwave irradiation. Tetrahedron Lett 52:2808–2811
Rovero P (2000) Homodetic cyclic peptides. In: Kates SA, Albericio F (eds) Solid-phase synthesis. A practical guide. Marcel Dekker, New York, NY, pp 331–364
Rovero P, Quartara L, Fabbri G (1991) Synthesis of cyclic peptides on solid support. Tetrahedron Lett 32:2639–2642
Scognamiglio PL, Di Natale C, Perretta G, Marasco D (2013) Peptides to small molecules: an intriguing but intricated way to new drugs. Curr Med Chem 20:3803–3817
Teixido M, Altamura M, Quartara L, Giolitti A, Maggi CA, Giralt E, Albericio F (2003) Bicyclic homodetic peptide libraries: comparison of synthetic strategies for their solid-phase synthesis. J Comb Chem 5:760–768
Thieriet N, Guibé F, Albericio F (2000) Solid-phase peptide synthesis in the reverse direction. Org Lett 2:1815–1817
Torres-Garcia C, Diaz M, Blasi D, Farras I, Fernandez I, Ariza X, Farras J, Lloyd-Williams P, Royo M, Nicolas E (2012a) Side-chain anchoring of tryptophan to solid supports using a dihydropyranyl handle: synthesis of brevianamide F. Int J Pept Res Ther 18:7–19
Torres-Garcia C, Pulido D, Carceller M, Ramos I, Royo M, Nicolas E (2012b) Solid-phase synthesis of phenylalanine containing peptides using a traceless triazene linker. J Org Chem 77:9852–9858
Trzeciak A, Bannwarth W (1992) Synthesis of ‘head-to-tail’ cyclized peptides on solid support by Fmoc chemistry. Tetrahedron Lett 33:4557–4560
Tulla-Puche J, Getun IV, Alsina J, Albericio F, Barany G (2004) Synthetic circularized analogues of bovine pancreatic trypsin inhibitor. Eur J Org Chem 4541–4544
Udenigwe CC, Howard A (2013) Meat proteome as source of functional biopeptides. Food Res Int 54:1021–1032
Vasconcelos L, Paern K, Langel U (2013) Therapeutic potential of cell-penetrating peptides. Therapy Deliv 4:573–591
Villorbina G, Canals D, Carde L, Grijalvo S, Pascual R, Rabal O, Teixidó J, Fabriàs G, Llebaria A, Casas J, Delgado A (2007) Solid-phase synthesis of a combinatorial library of dihydroceramide analogues and its activity in human alveolar epithelial cells. Bioorg Med Chem 15:50–62
Yan LZ, Edwards P, Flora D, Mayer JP (2004) Synthesis of cyclic peptides through hydroxyl side-chain anchoring. Tetrahedron Lett 45:923–925
Yraola F, Ventura R, Vendrell M, Colombo A, Fernandez J-C, de la Figuera N, Fernandez-Forner D, Royo M, Forns P, Albericio F (2004) A revaluation on the use of Rink, BAL, and PAL resins and linkers. QSAR Comb Sci 23:145–152
Zhong HM, Greco MN, Maryanoff BE (1997) Solid-phase synthesis of arginine-containing peptides by guanidine attachment to a sulfonyl linker. J Org Chem 62:9326–9330
Ziovas M, Tataraki D, Manousou P, Parveri N, Satoglou F, Gatos D Barlos K (2012) Solid phase peptide synthesis via side-chain attachment on resins of trityl and benzhydryl type. P210. 32 European peptide symposium, Athenes (Greece), 2–7 September
Zompra AA, Galanis AS, Werbitzky O, Albericio F (2009) Manufacturing peptides as active pharmaceutical ingredients. Future Med Chem 1:361–377
Acknowledgments
The work was partially supported by UKZN and NRF (South Africa) and CICYT (CTQ2012-30930), the Generalitat de Catalunya (2009SGR 1024), and the Institute for Research in Biomedicine (Spain).
Conflict of interest
Authors declare that they do not have any financial conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Cherkupally, P., Acosta, G.A., Ramesh, S. et al. Solid-phase peptide synthesis (SPPS), C-terminal vs. side-chain anchoring: a reality or a myth. Amino Acids 46, 1827–1838 (2014). https://doi.org/10.1007/s00726-014-1746-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-014-1746-7