
Abstract
Epibrassinolide (EBR) is a biologically active compound of the brassinosteroids, steroid-derived plant growth regulator family. Generally, brassinosteroids are known for their cell expansion and cell division-promoting roles. Recently, EBR was shown as a potential apoptotic inducer in various cancer cells without affecting the non-tumor cell growth. Androgen signaling controls cell proliferation through the interaction with the androgen receptor (AR) in the prostate gland. Initially, the development of prostate cancer is driven by androgens. However, in later stages, a progress to the androgen-independent stage is observed, resulting in metastatic prostate cancer. The androgen-responsive or -irresponsive cells are responsible for tumor heterogeneity, which is an obstacle to effective anti-cancer therapy. Polyamines are amine-derived organic compounds, known for their role in abnormal cell proliferation as well as during malignant transformation. Polyamine catabolism-targeting agents are being investigated against human cancers. Many chemotherapeutic agents including polyamine analogs have been demonstrated to induce polyamine catabolism that depletes polyamine levels and causes apoptosis in tumor models. In our study, we aimed to investigate the mechanism of apoptotic cell death induced by EBR, related with polyamine biosynthetic and catabolic pathways in LNCaP (AR+), DU145 (AR−) prostate cancer cell lines and PNT1a normal prostate epithelial cell line. Induction of apoptotic cell death was observed in prostate cancer cell lines after EBR treatment. In addition, EBR induced the decrease of intracellular polyamine levels, accompanied by a significant ornithine decarboxylase (ODC) down-regulation in each prostate cancer cell and also modulated ODC antizyme and antizyme inhibitor expression levels only in LNCaP cells. Catabolic enzymes SSAT and PAO expression levels were up-regulated in both cell lines; however, the specific SSAT and PAO siRNA treatments prevented the EBR-induced apoptosis only in LNCaP (AR+) cells. In a similar way, MDL 72,527, the specific PAO and SMO inhibitor, co-treatment with EBR during 24 h, reduced the formation of cleaved fragments of PARP in LNCaP (AR+) cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brassinosteroids (BRs) are steroid-derived plant hormones with growth-promoting roles in plants. A transmembrane heterodimer receptor, brassinosteroid insensitive 1 (BRI1) and BRI1-associated receptor kinase (BAK1) were found to be responsible for the BR-signaling mechanism (Bishop and Koncz 2002; Li et al. 2002). This receptor complex acts like mammalian serine/threonine kinases and triggers transcription of several genes responsible for proliferation and cell cycle control (Hu et al. 2000; Nam and Li 2002). Epibrassinolide (EBR), a natural BR derivative, was shown to be a plant regulator with a similar structure to mammalian steroids (Ali et al. 2008). Besides its well-known cell regulator function in plants, recently the effects of EBR on human diseases have been elucidated (Malikova et al. 2008; Carange et al. 2011). EBR has been proposed as an important molecule to treat neurodegenerative, steroid-induced disorders and cancer, namely diseases characterized by increased cell proliferation (Romanutti et al. 2007; Steigerova et al. 2010; Carange et al. 2011). The antiproliferative effect of EBR was effective in a dose-dependent manner through the induction of cell cycle arrest in different cancer lines (Malikova et al. 2008; Steigerova et al. 2010). Nuclear hormone receptor expressing breast and prostate cancer cells were found to be more sensitive against EBR treatment than normal fibroblasts or hormone independently growing malign cell lines (Wu and Lou 2007; Malikova et al. 2008; Steigerova et al. 2010).
Prostate cancer development generally depends on the ratio of proliferating cells versus apoptotic cells in the prostate tissue. Mainly, androgens positively regulate this balance (Taplin and Ho 2001). Although androgen ablation is one of the effective therapy models against prostate cancer, the transition from the androgen-dependent to a castration-resistant tumor phenotype is one of the obstacles. The prostate gland has among the highest level of polyamines (PAs) in the body (Schipper et al. 2003). PAs are multifunctional polycations found in all living organisms and are responsible for prostatic growth and differentiation. The biological behavior of prostate cancer cells has been shown to be associated with changes in PA levels and/or the activity of their metabolic enzymes (Seiler et al. 1996). PAs are also involved in many biochemical processes including cellular proliferation, cell cycle regulation and protein synthesis (Gerner and Meyskens 2004). Due to their proliferation-promoting roles, they are tightly regulated in mammalian cells. The PA biosynthetic enzyme ODC activity is controlled by OAZ and AZI (Su et al. 2009).This regulation was found to be disrupted in cancer cells (Wang et al. 2007; Kahana 2009). Since elevated PA levels are increased in rapidly proliferating cancer cells compared to healthy tissues, induction of PA catabolism provides an attractive therapeutic approach for many cancers (Devereux et al. 2003; Hector et al. 2008; Nowotarski et al. 2013). PA catabolism is a two-step process. Following the acetylation of PAs by spermidine/spermine N 1-acetyltransferase (SSAT), acetylated compounds are oxidized by PA oxidase (PAO). This process induces an impairment of intracellular PA content and production of toxic by-products such as hydrogen peroxide and aldehydes, which are strong apoptotic inducers (Agostinelli et al. 2004; Allen et al. 2007).
In this study, we investigated the potential effect of EBR in inducing apoptosis in androgen-responsive LNCaP and irresponsive DU145 cells in relation with PA catabolism. Our data indicated that EBR activated mitochondria-mediated and caspase-dependent apoptosis by modulating Bcl-2 family members in both cell lines. Additionally, catabolic enzymes of PAs were involved in EBR-induced apoptosis, more significantly in LNCaP androgen-responsive cell line than DU145 cells, suggesting that the AR status and PA catabolism are determinative in EBR-induced apoptosis.
Materials and methods
Chemicals, antibodies and primers
EBR and MDL 72,527 (PAO inhibitor, 10 mm stock concentration dissolved in DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). PARP, Puma, Bak, Bax, Bik, Bcl-xS,Mcl-1, Bcl-2,Bcl-xL, cleaved caspase-9, cleaved caspase-3, cleaved caspase 7, β-actin, GAPDH primary anti-rabbit antibodies (all employed 1:1,000), and horseradish peroxidase (HRP)-conjugated secondary anti-rabbit and anti-mouse antibodies (1:5,000) were from Cell Signaling Technology (CST, Danvers, MA, USA). Cytochrome c and Apaf-1 anti-rabbit antibodies (1:1,000) were purchased from BD Biosciences (San Jose, CA, USA). ODC, SSAT and PAO anti-rabbit antibodies (1:1,000), siRNA and transfection reagent were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). OAZ and AZI anti-mouse antibodies (1:1,000) were gifts from Dr. Chaim Kahana from Weizmann Institute, Israel. The appropriate primers for quantitative RT-PCR experiments were purchased from Sigma-Aldrich and prepared according to the manufacturer’s instructions.
Cell culture
LNCaP (CRL-1740) and DU145 (CRL-2698) human prostate cancer cell lines were obtained from American Type Culture Collection (ATCC). PNT1a normal prostate epithelial cell line was purchased from European Collection of Cell Culture (ECACC). Cells were grown in RPMI 1640 medium (Gibco; Invitrogen, Carlsbad, CA, USA), supplemented with 10 % fetal bovine serum (PAN Biotech, Aidenbach, Germany), 10,000 U penicillin/ml and 10 mg streptomycin/ml (PAN Biotech). Cells were cultured at 37 °C in a humidified 5 % CO2 incubator (HERA cell 150; Thermo Electron Corporation, Waltham, MA, USA). Cells were seeded overnight and then treated with the desired drug concentrations.
Cell viability assay
Cells were seeded at 1 × 104 density per well in 96-well plates and exposed to various concentrations of EBR for 24 and 48 h. 10 μl 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) dye (5 mg/ml) (Sigma-Aldrich) was added to each well and cells were kept at 37 °C for 4 h. The resulting formazan crystals were solubilized in 200 μl DMSO. The density of the solubilized formazan was read at 570 nm spectrophotometrically (Bio-Rad, Hercules, CA, USA).
Trypan blue dye exclusion assay
Cells were seeded in six-well plates (5 × 104 cells/well) and treated with EBR (25 μM) for 96 h. Every 24 h, cells were trypsinized and stained with trypan blue, and viable and dead cells were counted under light microscopy. The number of viable cells (y-axis) was plotted against time (x-axis).
Cell cycle analysis
Each cell line was seeded at a density of 2 × 105 cells/well in six-well plates and then treated with EBR (25 μM) for 24 h. Both floating and adherent cells were collected and fixed for 30 min with cold ethanol (70 % solution in distilled H2O). After centrifugation, pellets were resuspended in 1× PBS containing RNase (100 μg/ml) (Fermentas, St. Leon-Rot, Germany) and propidium iodide (PI) (Applichem, Darmstadt, Germany) solution (40 μg/ml). Samples were kept for 30 min at 37 °C in dark and then analyzed on an FACScan flow cytometer (Becton–Dickinson, San Jose, CA, USA), equipped with a 15 mW, 488 nm, air-cooled argon ion laser. Fluorescence emission was collected through a 570 nm band-pass filter. Cell cycle distribution was performed by CellQuest software (Becton-Dickinson, USA).
Evaluation of apoptotic cell death by Annexin V–FITC staining
To detect the phosphatidylserine translocation on the surface of plasma membrane (early marker of apoptosis induction), LNCaP, DU145 and PNT1a cells were seeded in six-well plates (3 × 105 cells/well) and then treated with 25 μM EBR for 24 h. Both floating and adherent cells were collected, resuspended in Annexin V binding buffer and then incubated with Annexin V–FITC and PI following the manufacturer’s instructions (MBL International, Woburn, MA, USA). 10,000 events per sample were acquired on the FACScan. Fluorescence emissions were collected through 530 and 570 nm band-pass filters for FITC and PI, respectively. Data are presented as dot plots (Annexin fluorescence on the x-axis; PI fluorescence on the y-axis). The numbers shown in the four quadrants represent the percentage of viable (lower left), necrotic (upper left), early apoptotic (lower right) and late apoptotic (upper right) cells evaluated using CellQuest software.
Apoptosis determination by cell death ELISA assay
Cytoplasmic histone-associated DNA fragments were determined according to the manufacturer’s instructions provided in Cell Death Detection ELISA Plus (Roche, Mannheim, Germany). 1 × 105 cells/well were seeded in 96-well plates, allowed to attach and then treated with 25 μM EBR for 24 h. The cell lysates were placed in streptavidin-coated microplate dishes. A mixture of anti-histone–biotin and anti-DNA–POD was added and incubated for 2 h at room temperature. After removal of unbound antibodies by a washing procedure, fragmented DNA was determined photometrically at 405 nm (Bio-Rad, Hercules, CA, USA).
Detection of mitochondrial membrane potential (Δψm) disruption
LNCaP and DU145 cells (1 × 105) were seeded into 12-well plates. Following exposure to EBR, cells were washed once with 1× PBS and then stained with a 4 nm 3,3′-dihexyloxacarbocyanine iodide [DiOC6(3)] (Calbiochem, La Jolla, CA, USA; 40 nm stock concentration in DMSO) fluorescent probe. Δψm disruption was measured by Fluoroskan Ascent Fluorometer (Thermo Labsystems, Beverly, MA, USA) (excitation/emission = 488/525 nm).
Immunoblot analysis
Cells were cultured in 60 mm Petri dishes in complete medium, then media were discarded and washed with ice-cold 1X PBS and lysed with ProteoJET Mammalian cell lysis buffer (Fermentas, St. Leon-Rot, Germany). Total protein levels were determined by Bradford method (Krauspe and Scheer 1986) (Bio-Rad, Hercules, CA, USA). Total cell lysates were separated by 12 %SDS-PAGE gels and electrotransferred onto polyvinylidene difluoride (PVDF) membranes (Roche, Mannheim, Germany) subjected to electrophoresis. Membranes were washed in tris-buffered saline with Tween 20 (TBS-T) [10 mm Tris–HCl (pH 8.3), 0.05 % Tween 20] (Tween 20, Sigma Ultra, St. Louis, MO, USA). Blocking was proceeded by 5 % skim milk containing TBS-T milk overnight at 4 °C. PVDF membranes were incubated with buffer containing 5 % (v:v) skim milk solution with appropriate antibodies. Membranes were rinsed with TBS-Tween 20 and incubated with HRP-conjugated secondary antibodies at 4 °C overnight. Following addition of enhanced chemiluminescence reagent (Lumi-Light Western blotting substrate) (Roche, Mannheim, Germany) membranes were exposed to X-ray films (Roche).
Measurement of PA levels
The PA content of each sample was determined by HPLC (Agilent Technologies, Wilmington, DE, USA) as described by Singh et al. (1992). Briefly, 1 × 106 cells were harvested from 60 mm Petri dishes, washed with 1× PBS and then pelletized. The cell pellet was solubilized in 5 % trichloroacetic acid and centrifuged at 13,200 rpm for 20 min. The supernatant was kept and benzoylation processes were performed. Endogenous PA levels were determined using high-pressure liquid chromatography (Agilent Technologies, Palo Alto, CA, USA).
Transient silencing of SSAT and PAO
Cells were seeded (1 × 106cells/well) into 60 mm Petri dishes. 1.25 μg/μl SSAT siRNA and PAO siRNA in the presence of 1:6 siRNA transfection reagent were prepared. The mixture was incubated for 15 min at room temperature and gently added dropwise onto cells in 3 ml of RPMI medium. After 48 h of siRNA transfection, cells were treated with 25 μM EBR and total RNA was isolated for SSAT and PAO mRNA expression analysis or protein extraction for western blotting.
RNA extraction and qRT-PCR
Total RNA was isolated by TRIzol (Invitrogen, USA) following the manufacturer’s instructions. cDNAs from each sample were obtained by using iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). The cDNAs were subjected to quantitative real-time PCR. Forty cycles of reactions were performed under the conditions of denaturation for 30 s at 95 °C, annealing was performed for 30 s at 55 °C and extension for 30 s at 72 °C. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal standard. PCRs were performed in separate tubes in duplicate in Mini-Opticon (Bio-Rad, Hercules, CA, USA). SSAT and PAO cDNAs were amplified using SSAT and PAO primers purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). GAPDH, internal standard cDNA, was also amplified using the following primers: forward, 5′-CGGAGTCAACGGATTTGGTCGTAT-3′; reverse, 5′-AGCCTTCTCCATGGTGGTGAAGAC-3′.
Determination of reactive oxygen species by DCFH-DA staining
Each prostate cancer cell line was seeded at a density of 1 × 104 cell/well in 96-well plates. Following exposure of cells with EBR for 24 h, media were carefully discarded and samples stained with 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (0.5 μM). Drug-induced reactive oxygen species (ROS) generation compared to untreated control samples was visualized by fluorescence microscopy (200X) (Olympus, Japan).
Statistical analysis
Results were analyzed with MS Office Excel 2007. Statistical significance test was utilized by using one-tailed unpaired t test. p < 0.05 was taken as a level of significance. Immunoblotting results were repeated at least twice and ImageJ program was used to measure band intensities.
Results and discussion
The effect of EBR on cell viability and survival of prostate cancer cells
Cells were incubated with various doses (0–100 μM) of EBR for 24 or 48 h and cell viability was determined by MTT assay. EBR induced cell viability loss in dose- and time-dependent manner compared to untreated samples in LNCaP and DU145 prostate cancer cells (Fig. 1a, b). Increasing concentrations of EBR was more effective on LNCaP cell viability loss than DU145 cells suggesting that androgen-dependent cells were more sensitive to EBR than androgen-independent prostate cancer cells. In further experiments, 25 μM EBR was selected due to its moderate cytotoxic effect on both cell lines. First, we examined the effect of EBR treatment on cell proliferation by counting the cell number within 96 h. A higher and earlier inhibition of cell proliferation was observed in LNCaP than DU145 cells (Fig. 1c, d). Similar to our finding, a previous report indicated that estrogen receptor-positive MCF-7 and estrogen receptor-negative MDA-MB-468 breast cancer cells might display different sensitivity against EBR treatment due to the functional estrogen receptor status (Steigerova et al. 2010). Therefore, we suggest that the presence of hormone receptors may be critical in EBR response.

EBR decreased cell viability in dose- and time-dependent manner in LNCaP and DU145 prostate cancer cells. a, b 1 × 104 cells were seeded into a 96-well plate and treated with EBR (0–100 μM) for 24 and 48 h. Cell viability loss was determined by MTT assay. Values represent the mean ± SD of three different experiments with at least five replicates. c, d Cells were seeded into a 96-well plate and treated with EBR (25 μM) for 24, 48, 72 and 96 h. Cell proliferation was determined by trypan blue dye exclusion method. Values represent the mean ± SD of three different experiments with at least five replicates
The effect of EBR on cell cycle distribution
Previous reports suggested that EBR caused significant cell cycle arrest at the G1 phase in various breast cancer cells (Steigerova et al. 2010) and led to apoptotic induction. However, in our experimental system, we did not observe a G1 block. As shown in Fig. 2a, EBR treatment (25 μM, 24 h) decreased the percentage of cells in the G1 phase by 2.5 and 1.4 fold in LNCaP and DU145 cells compared to untreated samples, respectively. The number of cells in the sub-G1 phase, which indicates cells with fragmented DNA, was increased in both cell lines (approximately, 49 % in LNCaP and 13 % in DU145 cells). In addition, no significant change in the sub-G1 phase was detected after EBR treatment in PNT1a normal prostate epithelial cells (Supplemental Figure 1a). Similar to this observation, Malikova et al. (2008) showed that BRs did not decrease cell viability in BJ fibroblast cells, suggesting that BRs induced different effects in normal and malign cell types.

EBR altered cell cycle distribution in LNCaP and DU145 cells. a, b LNCaP and DU145 cells were seeded into a six-well plate and treated with EBR (25 μM) for 24 h. After ethanol fixation and PI (propidium iodide) staining, samples were analyzed by flow cytometry. Cell cycle distribution of LNCaP and DU145 cells were determined by CellQuest software. Data shown are obtained from a representative experiment
To verify if EBR-induced cytotoxicity was due to apoptosis induction, Annexin V–FITC labeling was performed following 24 h of treatment. In DU145 cells, the percentage of early and late apoptotic cells increased by 7 % compared to untreated cells, while the apoptotic population was augmented by 22 % in LNCaP cells (Fig. 3a, b).

EBR induced apoptosis in LNCaP and DU145 prostate cancer cells. 3 × 105 cells were seeded into a six-well plate and treated with EBR for 24 h. Annexin V–PI staining was performed to determine apoptotic cell populations. a Untreated cells, b EBR treated (25 μM). Fluorescence signals from Annexin V–FITC and from PI are reported on the x-axis and y-axis, respectively. Numbers shown in the four quadrants represent the percentage of viable (lower left), necrotic (upper left), early apoptotic (lower right) and late apoptotic (upper right) cells. Data shown are obtained from a representative experiment
EBR treatment induced mitochondria-mediated apoptosis by activating caspase
Exposure of LNCaP and DU145 cells to 25 μM EBR for 24 h induced DNA fragmentation (3.5- and 2.3-fold, respectively) compared to untreated samples (***p < 0.0001) (Fig. 4a). Although EBR disrupted ΔψM in both cell lines as an apoptotic agent (Fig. 4b), cytochrome c release into cytosol was more remarkable in LNCaP than DU145cells (Fig. 4c). Apaf-1 and cleavage of caspase-9 were increased after EBR treatment in both cell lines. In addition, EBR induced cleavage of caspase-3 only in LNCaP cells (Fig. 4d), while cleaved caspase-7 was only induced in DU145 cells. PARP cleavage, a final step of caspase activation, was observed in both cell lines. These results suggest that EBR induced apoptosis by activating different caspases in a cell line-dependent manner. EBR treatment of PNT1a normal prostate epithelial cells did not induce significant alterations in Apaf-1, cleaved caspase-9 and PARP expressions (Supplemental Figure 1). Similar to our finding, EBR induced caspase-3 activation in MDA-MB-468 breast cancer cells (Steigerova et al. 2010). Genistein, a member of natural isoflavones and a nuclear receptor antagonist (Steigerova et al. 2010), induced apoptosis by activating caspases in prostate cancer cells (Kumi-Diaka et al. 2000). Especially, caspase-3 cleavage was required in LNCaP cells following genistein treatment. In our experimental model, the different cellular sensitivity against EBR might be a reason for activation of caspase-7 instead of caspase-3 in DU145 cells.

EBR-induced apoptosis was mitochondria-mediated and caspase dependent. a Cells were treated for 24 h with EBR, and DNA fragmentation was determined with cell death ELİSA. b Relative Δψm was evaluated after 24 h of EBR treatment with DiOC6 staining by fluorometric analysis (ex: 488 nm, em: 525 nm). c Following 24 h of EBR treatment, cytosolic proteins were isolated and separated in 12 % SDS gel, transferred onto PVDF membrane and blotted with anti-cytochrome c antibody. d Total proteins were isolated after EBR treatment and the expression profiles of Apaf-1, cleaved caspase-9, cleaved caspase-3, cleaved capase-7 and PARP were determined by immunoblotting using appropriate antibodies. β-actin was used as a loading control
EBR effectively modulated Bcl-2 family members
Recently, the effect of EBR on Bcl-2 and Bcl-xL down-regulation was established in breast cancer cell lines (Steigerova et al. 2012). Therefore, we investigated the apoptotic key players of Bcl-2 family members following EBR treatment (25 μM) for 24 h in LNCaP and DU145 prostate cancer cells. The expression of pro-apoptotic members (Puma α and β, Bak, Bax, Bik and Bcl-xS) and anti-apoptotic members (Mcl-1, Bcl-2 and Bcl-xL) were determined by western blotting method. Exposure to EBR significantly up-regulated pro-apoptotic Puma α and β, Bax, Bik and Bcl-xS expressions in LNCaP cells (Fig. 5). In DU145 cells while Puma α, β, Bax and Bak expressions were up-regulated, Bik and Bcl-xS expression profiles were not affected by EBR treatment (Fig. 5). EBR down-regulated anti-apoptotic protein Mcl-1 and Bcl-xL in LNCaP cells (Fig. 5). On the contrary, only Mcl-1 was down-regulated after EBR treatment and no significant change was observed in Bcl-xL expression levels in DU145 cells. The different modulation of pro- and anti-apoptotic proteins after EBR treatment might explain the different sensitivity and apoptotic response of LNCaP and DU145 against the drug.

EBR-modulated Bcl-2 family proteins expressions. The effect of EBR (25 μM) on Bcl-2 family proteins was determined in prostate cancer cells by immunoblotting. Following 24 h EBR treatment, total proteins were isolated and separated in 12 % SDS gel, transferred onto PVDF membranes and blotted with Puma, Bak, Bax, Bik, Bcl-xS, Bcl-2, Mcl-1, Bcl-2 and Bcl-xL antibodies. GAPDH was used as a loading control
Depletion of PAs by modulation of biosynthetic enzymes following EBR treatment
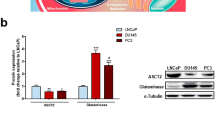
PA mimicking analogs might activate the catabolic enzymes of PAs by inducing depletion of intracellular PAs in different cancer cells (Allen et al. 2007; Hector et al. 2008). Similarly, we found that EBR treatment induced intracellular PAs depletion in prostate cancer cells. While Put and Spd levels were decreased by 50 % in LNCaP cells, EBR was able to decrease only Put level in DU145 cells (Fig. 6a). To understand the effect of EBR on intracellular PA pool, we investigated the biosynthetic key enzyme of PAs, ODC, by western blotting method. As shown in Fig. 6b, EBR treatment (25 μM, 24 h) caused AZI (the main inhibitor of OAZ) down-regulation in LNCaP and a slight increase in DU145 cells. However, OAZ expression was up-regulated after EBR treatment in LNCaP cells. Consequently, although EBR treatment decreased PA content in DU145 cells, it was not effective on expression profiles of ODC regulatory enzymes.

EBR depleted PAs by modulating PA metabolic enzymes in prostate cancer cells. a 1 × 106 cells were seeded onto six-well plates. Cells were treated with EBR for 24 h. PA content was determined by benzoylation method. Columns represent the mean ± SD from two experiments with at least two replicates. Put putrescine, Spd spermidine, Spm spermine. b ODC, OAZ and AZI. c SSAT and PAO expressions were determined by immunoblotting. Following 24 h EBR treatment, total proteins were isolated and separated in 12 % SDS gel, transferred onto a PVDF membrane and blotted with appropriate antibodies. β-actin was used as a loading control
It is well established that increased intracellular PAs are critical in different cellular signaling cascades in tumor cells (Agostinelli et al. 2009, 2010). Since PAs are key regulators of cell proliferation, depletion of PA content in cancer cells has gained attention in cancer therapy. Recent studies have indicated that chemotherapeutic agents such as cisplatin and doxorubicin could alter intracellular PA by disrupting the regulation of PA metabolic enzymes. Therefore, in our study, we determined the expression levels of PA biosynthetic and catabolic enzymes in response to EBR treatment. The down-regulation of ODC and AZI and up-regulation of OAZ might explain the decrease in Put content of LNCaP cells (Fig. 6a). In another aspect, the up-regulation of OAZ and down-regulation of AZI following EBR treatment might also suggest that the PA uptake mechanism besides ODC gene regulation was an important target for EBR in LNCaP cells. Conversely, although ODC expression was down-regulated, due to unaffected OAZ and AZI, PA uptake should still be present in DU145 cells. We hypothesize that intracellular Spd and Spm levels which are targets of catabolic enzymes of PAs are determinants of cellular apoptotic response to EBR. It is well established that knocking down AZI by shRNA caused the increase in OAZ ratio relative to AZI, thus promoting growth activation during neoplastic transformation. This finding was also confirmed by OAZ overexpression in human PC3 M-LN4 and rat AT6.1 prostate cancer cell lines (Olsen et al. 2012).
The catabolic activity of PA oxidases may trigger the generation of toxic by-products, such as H2O2 as an important apoptotic inducer. There are several reports showing that up-regulated SSAT and PAO gene expressions caused the accumulation of higher levels of ROS and apoptosis in breast and colon cancer cells lines (Arisan et al. 2012; Coker et al. 2012). Therefore, we investigated the modulation of PA catabolic enzymes SSAT and PAO expressions after EBR treatment in LNCaP and DU145 cells. As shown in Fig. 6c, EBR was able to up-regulate SSAT and PAO expression levels in both prostate cancer cells. Correspondingly, mRNA levels of these genes were checked only in drug-sensitive LNCaP cells by the qRT-PCR method. We found that EBR treatment caused up-regulation of SSAT and PAO by six and twofold compared to control samples, respectively (Supplemental Figure 2a, b). These findings showed that EBR activated PA catabolism by affecting the transcriptional and translational mechanisms in the cells.
Silencing of PAO and SSAT prevented EBR-induced apoptotic cell death in LNCaP cells
To demonstrate clearly the relation between EBR and ROS generation due to modulated PA catabolic activity, we transiently silenced SSAT or PAO in LNCaP cells (Supplemental Figure 2a, b). We found that silencing of SSAT or PAO significantly prevented EBR-induced cell viability loss in LNCaP cells. In contrast, under the same experimental conditions, silencing of SSAT or PAO did not prevent cell viability loss after EBR treatment in DU145 cells (Fig. 7a, c). For this reason in subsequent experiments, we only examined LNCaP cells to understand the relation between functional PA catabolic machinery and EBR treatment.

SSAT and PAO silencing prevented EBR-induced apoptosis. a, c Cell viability loss was determined by MTT assay after 100 nM SSAT or PAO silencing prior to 24 h EBR treatment. b, d DNA fragmentation profiles after SSAT and PAO silencing and EBR treatment were determined by cell death ELISA. e The reactive oxygen species generation was determined by DCFH-DA staining following 48 h of PAO silencing and 24 h of EBR treatment in LNCaP cells. NS non-silencing control siRNA
To understand how activated PA catabolic enzymes contributed to EBR-induced apoptosis in LNCaP cells in association with decreased cell viability, we investigated DNA laddering ratio. Silencing of SSAT or PAO for 48 h significantly prevented EBR-induced apoptosis in LNCaP cells (Fig. 7b, d). In addition, diminished PA catabolic enzyme activity prevented EBR-induced ROS accumulation in LNCaP cells (Fig. 7e). Therefore, these observations suggested that accumulation of ROS due to activation of PA catabolic enzymes after drug treatment caused apoptotic stimuli in LNCaP cells. We also confirmed this result by inhibiting PAO activity with MDL 72,527, the specific PAO/SMO inhibitor. In the presence of MDL 72,527, in a similar way with PAO silencing results, inhibition of PAO enzymatic activity prevented cell viability loss and apoptosis in LNCaP cells (Fig. 8a, b).

PAO was critical in EBR-induced apoptosis. a Cell viability loss following combined treatment of MDL 72,527 (50 μM) and EBR (25 μM) for 24 h was determined by MTT assay. b The effect of co-treatment of MDL 72,527 and EBR on EBR-induced apoptosis was determined by PARP immunoblotting. After 24 h, MDL 72,527 and EBR co-treatment total protein isolation was performed. Proteins were separated in 12 % SDS gel, transferred onto PVDF membrane and blotted with PARP antibody
It should be noted that the effect of the serum amine oxidases as a possible source of H2O2 was excluded by their inhibitor, amino guanidine. There are conflicting reports about MDL 72,527 effects on cancer cells. It was shown that MDL 72,527 alone could induce apoptosis by increasing the lysosomotropic effect in the cells (Agostinelli and Seiler 2007). However, the antagonistic effect of MDL 72,527 on PA analog N 1-ethyl-N 11-[(cycloheptyl) methy]-4,8-diazaundecane (CHENSpm) was observed in colon cancer cells (Lawson et al. 2002). Similar to this finding, MDL 72,527 induced anti-apoptotic effects in prostate cancer cells (Basu et al. 2009). Although all findings in this study pointed out that H2O2 was the primary source of apoptotic induction in an androgen receptor status-independent manner due to EBR treatment, inhibition of amine oxidases by MDL 72,527 or silencing of key catabolic enzymes of PAs prevented EBR-induced apoptosis. For this reason, we conclude that the functional PA metabolic machinery is critical in rapid response against EBR treatment only in tumor cells.
Conclusion
EBR induced mitochondria-mediated apoptosis in AR (+) LNCaP and AR (−) DU145 cells without affecting PNT1a normal prostate epithelial cells. The apoptotic efficiency of EBR was different in the two cell lines unrelated to the AR expression status. Our data indicated that the molecular determinants of intracellular PA levels might be critical to the EBR-induced apoptosis mechanism. All these findings could be important for the therapeutic potential of EBR in prostate cancer without damaging normal cells. Further studies should be addressed on PAs involvement in the EBR-induced cell death mechanism.
Abbreviations
- AR:
-
Androgen receptor
- AZI:
-
Antizyme inhibitor
- BAK1:
-
BRI1-associated receptor kinase
- BRI1:
-
Brassinosteroid insensitive 1
- BR:
-
Brassinosteroid
- DCFH-DA:
-
2′,7′-Dichlorofluorescein diacetate
- DiOC6:
-
3,3′-Dihexyloxacarbocyanine iodide
- DMSO:
-
Dimethyl sulfoxide
- EBR:
-
Epibrassinolide
- HPLC:
-
High-pressure liquid chromatography
- HRP:
-
Horseradish peroxidase
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide
- OAZ:
-
ODC antizyme
- ODC:
-
Ornithine decarboxylase
- PA:
-
Polyamine
- PAO:
-
Polyamine oxidase
- PBS:
-
Phosphate-buffered saline
- PVDF:
-
Polyvinyldifluoride
- ROS:
-
Reactive oxygen species
- SDS-PAGE:
-
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SSAT:
-
Spermidine/spermine N 1-acetyltransferase
- TBS-T:
-
Tris-buffered saline with Tween-20
- Δψm:
-
Mitochondrial membrane potential
References
Agostinelli E, Seiler N (2007) Lysosomotropic compounds and spermine enzymatic oxidation products in cancer therapy (review). Int J Oncol 31(3):473–484
Agostinelli E, Arancia G, Vedova LD, Belli F, Marra M, Salvi M, Toninello A (2004) The biological functions of polyamine oxidation products by amine oxidases: perspectives of clinical applications. Amino Acids 27(3–4):347–358
Agostinelli E, Condello M, Molinari A, Tempera G, Viceconte N, Arancia G (2009) Cytotoxicity of spermine oxidation products to multidrug resistant melanoma M14 ADR2 cells: sensitization by the MDL 72527 lysosomotropic compound. Int J Oncol 35(3):485–498
Agostinelli E, Tempera G, Viceconte N, Saccoccio S, Battaglia V, Grancara S, Toninello A, Stevanato R (2010) Potential anticancer application of polyamine oxidation products formed by amine oxidase: a new therapeutic approach. Amino Acids 38(2):353–368
Ali B, Hayat S, Fariduddin Q, Ahmad A (2008) 24-epibrassinolide protects against the stress generated by salinity and nickel in Brassica juncea. Chemosphere 72(9):1387–1392
Allen WL, McLean EG, Boyer J, McCulla A, Wilson PM, Coyle V, Longley DB, Casero RA Jr, Johnston PG (2007) The role of spermidine/spermine N 1-acetyltransferase in determining response to chemotherapeutic agents in colorectal cancer cells. Mol Cancer Ther 6(1):128–137
Arisan ED, Obakan P, Coker A, Palavan-Unsal N (2012) Inhibition of ornithine decarboxylase alters the roscovitine-induced mitochondrial-mediated apoptosis in MCF-7 breast cancer cells. Mol Med Rep 5(5):1323–1329
Basu HS, Thompson TA, Church DR, Clower CC, Mehraein-Ghomi F, Amlong CA, Martin CT, Woster PM, Lindstrom MJ, Wilding G (2009) A small molecule polyamine oxidase inhibitor blocks androgen-induced oxidative stress and delays prostate cancer progression in the transgenic adenocarcinoma of the mouse prostate model. Cancer Res 69(19):7689–7695
Bishop GJ, Koncz C (2002) Brassinosteroids and plant steroid hormone signaling. Plant Cell 14(Suppl):S97–S110
Carange J, Longpre F, Daoust B, Martinoli MG (2011) 24-epibrassinolide, a phytosterol from the brassinosteroid family, protects dopaminergic cells against MPP-induced oxidative stress and apoptosis. J Toxicol 2011:392859
Coker A, Arisan ED, Palavan-Unsal N (2012) Silencing of the polyamine catabolic key enzyme SSAT prevents CDK inhibitor-induced apoptosis in Caco-2 colon cancer cells. Mol Med Rep 5(4):1037–1042
Devereux W, Wang Y, Stewart TM, Hacker A, Smith R, Frydman B, Valasinas AL, Reddy VK, Marton LJ, Ward TD, Woster PM, Casero RA (2003) Induction of the PAOh1/SMO polyamine oxidase by polyamine analogues in human lung carcinoma cells. Cancer Chemother Pharmacol 52(5):383–390
Gerner EW, Meyskens FL Jr (2004) Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer 4(10):781–792
Hector S, Tummala R, Kisiel ND, Diegelman P, Vujcic S, Clark K, Fakih M, Kramer DL, Porter CW, Pendyala L (2008) Polyamine catabolism in colorectal cancer cells following treatment with oxaliplatin, 5-fluorouracil and N 1, N 11 diethylnorspermine. Cancer Chemother Pharmacol 62(3):517–527
Hu Y, Bao F, Li J (2000) Promotive effect of brassinosteroids on cell division involves a distinct CycD3-induction pathway in Arabidopsis. Plant J 24(5):693–701
Kahana C (2009) Antizyme and antizyme inhibitor, a regulatory tango. Cell Mol Life Sci 66(15):2479–2488
Krauspe R, Scheer A (1986) Coomassie brilliant blue G-250 dye-binding technique for determination of autolytic protein breakdown in Euglena gracilis and comparison to other methods of autolysis measurement. Anal Biochem 153(2):242–250
Kumi-Diaka J, Sanderson NA, Hall A (2000) The mediating role of caspase-3 protease in the intracellular mechanism of genistein-induced apoptosis in human prostatic carcinoma cell lines, DU145 and LNCaP. Biol Cell 92(8–9):595–604
Lawson KR, Marek S, Linehan JA, Woster PM, Casero RA Jr, Payne CM, Gerner EW (2002) Detoxification of the polyamine analogue N 1-ethyl-N 11-[(cycloheptyl)methy]-4,8-diazaundecane (CHENSpm) by polyamine oxidase. Clin Cancer Res 8(5):1241–1247
Li J, Wen J, Lease KA, Doke JT, Tax FE, Walker JC (2002) BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 110(2):213–222
Malikova J, Swaczynova J, Kolar Z, Strnad M (2008) Anticancer and antiproliferative activity of natural brassinosteroids. Phytochemistry 69(2):418–426
Nam KH, Li J (2002) BRI1/BAK1, a receptor kinase pair mediating brassinosteroid signaling. Cell 110(2):203–212
Nowotarski SL, Woster PM, Casero RA (2013) Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev Mol Med 15:e3
Olsen RR, Chung I, Zetter BR (2012) Knockdown of antizyme inhibitor decreases prostate tumor growth in vivo. Amino Acids 42(2–3):549–558
Romanutti C, Castilla V, Coto CE, Wachsman MB (2007) Antiviral effect of a synthetic brassinosteroid on the replication of vesicular stomatitis virus in Vero cells. Int J Antimicrob Agents 29(3):311–316
Schipper RG, Romijn JC, Cuijpers VM, Verhofstad AA (2003) Polyamines and prostatic cancer. Biochem Soc Trans 31(2):375–380
Seiler N, Delcros JG, Moulinoux JP (1996) Polyamine transport in mammalian cells. An update. Int J Biochem Cell Biol 28(8):843–861
Singh AB, Thomas TJ, Thomas T, Singh M, Mann RA (1992) Differential effects of polyamine homologues on the prevention of DL-alpha-difluoromethylornithine-mediated inhibition of malignant cell growth and normal immune response. Cancer Res 52(7):1840–1847
Steigerova J, Oklestkova J, Levkova M, Rarova L, Kolar Z, Strnad M (2010) Brassinosteroids cause cell cycle arrest and apoptosis of human breast cancer cells. Chem Biol Interact 188(3):487–496
Steigerova J, Rarova L, Oklest’kova J, Krizova K, Levkova M, Svachova M, Kolar Z, Strnad M (2012) Mechanisms of natural brassinosteroid-induced apoptosis of prostate cancer cells. Food Chem Toxicol 50(11):4068–4076
Su KL, Liao YF, Hung HC, Liu GY (2009) Critical factors determining dimerization of human antizyme inhibitor. J Biol Chem 284(39):26768–26777
Taplin ME, Ho SM (2001) Clinical review 134: the endocrinology of prostate cancer. J Clin Endocrinol Metab 86(8):3467–3477
Wang X, Feith DJ, Welsh P, Coleman CS, Lopez C, Woster PM, O’Brien TG, Pegg AE (2007) Studies of the mechanism by which increased spermidine/spermine N 1-acetyltransferase activity increases susceptibility to skin carcinogenesis. Carcinogenesis 28(11):2404–2411
Wu YD, Lou YJ (2007) Brassinolide, a plant sterol from pollen of Brassica napus L., induces apoptosis in human prostate cancer PC-3 cells. Pharmazie 62(5):392–395
Acknowledgments
This work was partially supported by Istanbul University Research Projects Center (Project no: 20958), Istanbul Kultur University Scientific Projects Support Center; Italian MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca) (EA); Istituto Superiore di Sanità “Project Italy–USA” (EA); funds MIUR-PRIN (Cofin) (EA) and Istituto Pasteur Fondazione Cenci Bolognetti (EA).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
726_2013_1574_MOESM1_ESM.pdf
EBR did not modulate cell cycle distribution and induce apoptosis in PNT1a prostate epithelial cells (a) PNT1a cells were seeded into a 6-well plate and treated with EBR (25 μM) for 24 h. After ethanol fixation and PI (propidium iodide) staining, samples were analyzed by flow cytometry. Cell cycle distribution of was determined by CellQuest software. Data shown are obtained from a representative experiment (b) Total proteins were isolated from PNT1a cells; the expression profiles of Apaf-1 and cleaved caspase-9 were determined by immunoblotting following 24 h EBR treatment while PARP expression was analyzed at 24, 48 and 72 h. β-actin was used as a loading control. (PDF 102 kb)
726_2013_1574_MOESM2_ESM.pdf
SSAT and PAO silencing determination with qRT-PCR. Cells were seeded in 6 well plates and treated with EBR for 24 h. Total RNA was isolated and cDNA synthesis was perfomed. To determine the efficiency of selected concentration of siRNA to silence (a) SSAT or (b) PAO, qRT-PCR with appropriate primers was performed. (PDF 62 kb)
Rights and permissions
About this article
Cite this article
Obakan, P., Arisan, E.D., Calcabrini, A. et al. Activation of polyamine catabolic enzymes involved in diverse responses against epibrassinolide-induced apoptosis in LNCaP and DU145 prostate cancer cell lines. Amino Acids 46, 553–564 (2014). https://doi.org/10.1007/s00726-013-1574-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-013-1574-1