
Abstract
Purpose
Resistance to androgen-deprivation therapies and progression to so-called castrate-resistant prostate cancer (CRPC) remain challenges in prostate cancer (PCa) management and treatment. Among other alterations, CRPC has been associated with metabolic reprogramming driven by androgens. Here, we investigated the role of androgens in regulating glutaminolysis in PCa cells and determined the relevance of this metabolic route in controlling the survival and growth of androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells.
Methods
PCa cells (LNCaP, DU145 and PC3) and 3-month old rats were treated with 5α-dihydrotestosterone (DHT). Alternatively, LNCaP cells were exposed to the glutaminase inhibitor BPTES, alone or in combination with the anti-androgen bicalutamide. Biochemical, Western blot and extracellular flux assays were used to evaluate the viability, proliferation, migration and metabolism of PCa cells in response to DHT treatment or glutaminase inhibition.
Results
We found that DHT up-regulated the expression of the glutamine transporter ASCT2 and glutaminase, both in vitro in LNCaP cells and in vivo in rat prostate cells. BPTES diminished the viability and migration of PCa cells, while increasing caspase-3 activity. CRPC cells were found to be more dependent on glutamine and more sensitive to glutaminase inhibition. BPTES and bicalutamide co-treatment had an additive effect on suppressing LNCaP cell viability. Finally, we found that inhibition of glutaminolysis differentially affected glycolysis and lipid metabolism in both androgen-sensitive and CRPC cells.
Conclusion
Our data reveal glutaminolysis as a central metabolic route controlling PCa cell fate and highlight the relevance of targeting glutaminase for CRPC treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Prostate cancer (PCa) is one of the most frequently diagnosed cancers among men. Its treatment and monitoring remain clinical challenges. PCa progression initially depends on the stimulatory action of androgens, which validates the use of therapies reducing androgen biosynthesis or antagonizing the androgen receptor (AR) [33, 71]. However, the majority of PCa cases becomes resistant to androgen-deprivation therapy (ADT) and progresses to so-called castrate-resistant PCa (CRPC), on average after 38 months [44]. This stage of the disease is characterized by the proliferation of PCa cells independent of androgen depletion and, despite important advances that have been made in recent years, effective treatment options for CRPC are still needed [25, 42]. Preclinical and clinical studies aimed at developing strategies for a better management of PCa and slowing down the progression to CRPC involved investigation of the efficacy of ADT in combination with other drugs, i.e., docetaxel or cabazitaxel [76]. As yet, however, the benefits are modest, increasing the overall survival time by a few months only, while development of resistance may occur [76]. The identification of new therapeutic targets related to the actions of androgens and the AR remains a fundamental aspect for improving the management and treatment of PCa.
Metabolic reprogramming favouring cancer development and progression is one of the hallmarks of cancer that has been intensively exploited for therapeutic improvement in recent years [13, 39, 51]. PCa progression to CRPC has been associated with a set of metabolic alterations that may be driven by androgens [52, 79, 80]. These androgens have been characterized as master regulators of several metabolic pathways in PCa, including aerobic glycolysis, glutamine metabolism, pyruvate import, oxidative phosphorylation, fatty acid β-oxidation and de novo lipid synthesis [4, 32, 80]. Indeed, their role in stimulating glucose and lipid usage and metabolism has been well established, but their capacity to modulate other routes, such as glutaminolysis, is less well known.
Glutamine is one of the principal energy sources, supplying nitrogen for purine and pyrimidine biosynthesis, as well as a carbon substrate for anabolic processes in cancer [16, 78]. Glutamine uptake is mediated by the alanine, serine, cysteine transporter 2 (ASCT2) (also called SLC1A5 or L-type amino acid transporter) whose expression has been shown to be significantly increased in several types of cancer, including PCa [50]. In the intracellular space, glutamine is converted to glutamate by the mitochondrial enzyme glutaminase [30]. Glutamate, in turn, generates α-ketoglutarate, an intermediate metabolite that fuels the tricarboxylic acid (TCA) cycle. In addition, glutaminase and α-ketoglutarate have been shown to influence other metabolic pathways, such as the glycolytic pathway [61].
Glutaminase has been shown to be highly expressed in PCa and to correlate positively with tumour stage and disease progression [61, 94]. Also, there are indications that glutaminolysis may represent a relevant metabolic pathway in PCa, and that inhibiting glutaminase can suppress PCa cell proliferation [9, 15, 26, 57, 64, 77]. However, the broad actions of androgens regulating this metabolic route and the effect of glutaminase inhibition in PCa cells in the presence (or absence) of anti-androgens still needs to be resolved. Also, it is unknown whether inhibition of glutaminolysis by blocking glutaminase activity can shape PCa cells towards higher glucose and lipid utilization.
The main objective of this study was to assess the importance of glutaminolysis in controlling the fate of androgen-sensitive and CRPC cells, and to investigate the androgenic regulation of this metabolic route. We also ascertained how PCa cells respond by using other metabolic pathways, such as glycolysis, after blockage of glutaminolysis.
2 Materials and methods
2.1 Chemicals
All chemicals, culture media and antibodies, unless otherwise stated, were purchased from Sigma-Aldrich (St Louis, MO, USA). All solutions were prepared with ultra-pure water. BPTES was dissolved in DMSO, and 5α-dihydrotestosterone (DHT) and bicalutamide in ethanol.
2.2 Cell lines and treatment
Human prostate adenocarcinoma cell lines (LNCaP, DU145 and PC3) were purchased from the European Collection of Cell Cultures (ECACC, Salisbury, UK). LNCaP cells originate from a PCa lymph node metastasis, express the AR and are used as an androgen-sensitive model. The AR-negative DU145 and PC3 cells originate from brain and bone metastases of undifferentiated grade IV prostate adenocarcinomas, respectively, and are used to mimic the castrate-resistant stage of PCa [45, 74].
LNCaP, DU145 and PC3 cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/ streptomycin at 37 °C in an atmosphere equilibrated with 5% CO2. At 60% confluence, the culture medium was replaced by phenol red-free RPMI-1640 medium containing 5% charcoal-stripped FBS. Next, the cells were maintained for an additional 24 h and subsequently exposed to DHT (10 nM) or vehicle for 12, 24 and 48 h. Alternatively, cells were exposed to different concentrations of glutamine (0, 0.25, 0.5, 0.75, 1 and 2 mM), or the glutaminase inhibitor BPTES (1, 2, 5, 10, 25 and 50 μM), alone or in combination with the AR inhibitor bicalutamide (10, 20 and 40 μM).
2.3 Animals and hormone treatment
Adult male Wistar (Rattus norvegicus) rats, aged ~3 months (250–300 g) were maintained in an animal room at constant temperature and humidity, under a 12 h light-dark cycle, with standard food pellets and water available ad libitum. Animal handling and all experimental procedures complied with the guidelines established by the “Guide for the Care and Use of Laboratory Animals” published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996), and the National and European Union rules for the care and handling of laboratory animals (Directive 2010/63/EU).
In total 16 rats were orchidectomised (ORCHX) under anaesthesia (Clorketam 1000, Vétoquinol, Lure, France). Five days after surgery, the animals were randomly divided into two groups receiving daily intraperitoneal injections of DHT (500 μg/kg/day) (ORCHX + DHT) or vehicle alone (physiologic serum/ethanol 30% alone; ORCHX + Vehicle) for 5 days. Another group of animals that did not undergo surgery was treated daily with vehicle alone (intact group, n = 8). After treatment, the animals were euthanized by cervical dislocation under anesthesia (100 mg ketamine/8 mg xilazine per Kg), after which the prostates were removed and immediately frozen in liquid nitrogen for protein extraction.
2.4 Protein extraction
Human prostate cells and rat prostate tissues were homogenized in appropriate volumes of radioimmunoprecipitation assay buffer (RIPA) (150 mM NaCl, 1% Nonidet-P40 substitute, 0.5% Na-deoxycholate, 0.1% SDS, 50 mM Tris, 1 mM EDTA) supplemented with a 1% protease inhibitors cocktail and PMSF (0,1 mM), kept on ice for 20 min with occasional mixing, and then centrifuged at 14,000 rpm for 20 min at 4 °C. Protein concentrations were determined using a Pierce™ BCA Protein Assay Kit (Thermo Scientific) according to the manufacturer’s instructions.
2.5 Western blot analysis
Twenty-five microgram of protein extracts were resolved on a 12.5% acryl-bisacrylamide gel by SDS-PAGE. Next, the proteins were electro-transferred to PVDF membranes (Bio-Rad, Hercules, CA, USA), which were subsequently incubated with rabbit anti-ASCT2 (1:1000, V501; Cell Signalling Technology, Danvers, MA, USA), rabbit anti-glutaminase (1:1000, ab93434, Abcam, Cambridge, UK), rabbit anti-glucose transporter type1 (GLUT1, 1:1000,CBL242, Millipore), rabbit anti-GLUT2 (1:1000, sc-9117, Santa Cruz Biotechnology, Heidelberg, Germany), rabbit anti-GLUT3 (1:1000, sc-30,107, Santa Cruz Biotechnology), rabbit anti-phosphofructokinase 1 (PFK1, 1:1000, sc-67,028, Santa Cruz Biotechnology), rabbit anti-lactate dehydrogenase (LDH, 1:10,000, Ab52488, Abcam,), rabbit anti-monocarboxylate transporter 4 (MCT4, 1:1000, sc-50,329,Santa Cruz Biotechnology), rabbit anti-fatty acid synthase (FASN, 1:1000, no.3180; Cell Signaling Technology), mouse anti-carnitine palmitoyltransferase 1A (CPT1A, 1:1000, ab128568; Abcam), rabbit anti-p21 (1:500, sc-397, Santa Cruz Biotechnology) and rabbit anti-phospho-c-Myc (1:1000, no.13748; Cell Signaling Technology) primary antibodies. Next, the membranes were washed and incubated with either anti-rabbit IgG HRP-linked (1:20000, sc-2004, Santa Cruz Biotechnology) or anti-mouse-IgGκ HRP-linked (1:20000, sc-516,102, Santa Cruz Biotechnology) secondary antibodies for 1 h at room temperature. Protein expression levels were normalized using an anti-α-tubulin monoclonal antibody (1:10000, T9026) or a mouse anti-β-actin antibody (1:1000, A5441). The membranes were incubated with ECL substrate (Bio-Rad) for 5 min, after which immunoreactive proteins were visualized using a ChemiDoc™ MP System (Bio-Rad). Band densities were obtained according to standard methods using Image Lab software (Bio-Rad) and normalized using the respective housekeeping band densities.
2.6 Fluorescence immunocytochemistry
LNCaP cells were fixed with 4% paraformaldehyde (PFA) for 10 min and permeabilized with 1% Triton X-100 for 5 min at room temperature. Next, non-specific staining was blocked by incubation with PBS containing 0.1% (w/v) Tween-20 (PBS-T) and 20% FBS for 1 h. After washing, cells were incubated with a rabbit anti-ASCT2 (1:100, V501; Cell Signalling Technology) primary antibody for 1 h at room temperature. Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen, Darmstadt, Germany) was used as secondary antibody. Alternatively, cells were incubated with a rabbit anti-calnexin (1:50, sc-11397, Santa Cruz Biotechnology) primary antibody for 1 h at room temperature after which Alexa Fluor 546 goat anti-rabbit IgG (Invitrogen) was used as secondary antibody. Specificity of staining was assessed by omission of the primary antibodies. Cell nuclei were stained with Hoechst 33342 (5 mg/ml, Invitrogen) for 10 min. Lamellae were washed with PBS-T and mounted using Dako fluorescent mounting medium (Dako, Glostrup, Denmark). Images were acquired using a Zeiss LSM710 laser scanning confocal microscope (Carl Zeiss, Gottingen, Germany). The threshold for co-localization of ASCT2 and calnexin was defined through omission of anti-calnexin or anti-ASCT2 antibodies. The degree of co-localization was measured using the Pearson’s correlation coefficient.
2.7 Cell viability assay
LNCaP (25.000 cells/well), DU145 (8000 cells/well) and PC3 (8000 cells/well) cells were seeded in 96-well plates, after which cell viability was determined using a colorimetric MTT assay. After BPTES or bicalutamide treatment, MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide), at a final concentration of 0.5 mg/ml, was added to the cell culture medium and incubated in the dark at 37 °C for 4 h. Next, the MTT solution was carefully removed, and the formed formazan crystals were solubilized using 100 μl DMSO. The absorbance of the resultant purple coloured solution was measured at 570 nm using a xMark™ Microplate Absorbance Spectrophotometer (Bio-Rad). The absorbance measured is directly proportional to the number of viable cells in each experimental group.
2.8 Cell migration assay
Cell migration assays were performed using 8 μM pore size transwell inserts (35,224, SPL, Life Sciences, Naechon-Myeon Pocheon, South Korea). Briefly, LNCaP (3.0 × 105 cells/transwell), DU145 (105 cells/transwell) and PC3 (1.5 × 105 cells/transwell) cells were seeded into the upper chambers with serum-free media in the presence or absence of BPTES. The lower chambers contained media supplemented with 20% FBS. After 24 h, cells on the lower surface of the transwell inserts were fixed and stained with haematoxylin. Next, the cells were counted in 10 randomly selected, 40× magnification fields per transwell.
2.9 Caspase-3-like activity assay
Caspase-3-like activity was determined spectrophotometrically at 405 nm by detecting the yellow product p-nitro-aniline (pNA) upon cleavage of caspase-3 substrate (Ac-DEVD-pNA). In brief, 10 μl total protein extract was incubated overnight at 37 °C with reaction buffer (20 mM HEPES, pH 7.4, 2 mM EDTA, 0.1% CHAPS, and 5 mM DTT) and 200 μM Ac-DEVD-pNA. The amount of generated pNA was calculated by extrapolation using a standard curve with known amounts of pNA. All measurements were normalized to the total amount (μg) of protein in each sample.
2.10 Glutamine, glucose and lactate quantification
The concentrations of glutamine (NZYTech, Lisboa, Portugal), glucose and lactate (Spinreact, Girona, Spain) in the cell culture media of DHT- or BPTES-treated cells were determined by spectrophotometric analysis using commercial kits according to the respective manufacturer’s instructions. Glutamine and glucose consumption, as well as lactate production, were calculated relative to the initial concentration of these metabolites at 0 h of treatment. All measurements were normalized to the total amount (μg) of protein in each sample.
2.11 Glycolytic rate assay
The cell glycolytic response upon BPTES treatment was evaluated by measurement of the extracellular acidification (proton efflux rate) using a Seahorse XF-96 analyser (Seahorse Bioscience, Agilent, Santa Clara, CA, USA). LNCaP (15,000 cells/well), DU145 (5000 cells/well) and PC3 (5000 cells/well) cells were seeded in 96-well plates (Seahorse Bioscience) and maintained at 37 °C, 5% CO2 for 24 h. Next, the cells were exposed to 10 μM BPTES for 24 h. In parallel, a XFe96 sensor cartridge for each cell plate was placed in a 96-well calibration plate containing 200 μl/well calibration buffer and left to hydrate overnight at 37 °C. The sensor cartridge and the calibration plate were loaded into a XFe96 Extracellular Flux Analyzer for calibration, after which the utility plate containing the calibration solution was replaced by the study plate, and the assay started. After 24 h of treatment with BPTES, the cell culture medium was replaced by 180 μl/well low-buffered serum-free medium without phenol red containing 2 mM glutamine, 10 mM glucose, 1 mM pyruvate and 5.0 mM HEPES at pH 7.4, and incubated at 37 °C for 1 h in the absence of CO2. The mitochondrial inhibitors rotenone and antimycin A (0.5 μM each) and glycolysis inhibitor 2-deoxy-D-glucose (2-DG, 50 mM) were injected via ports A and B, respectively.
The data were analysed using an Agilent Seahorse Glycolytic Rate Assay Report Generator, and the raw data were used to calculate the initial proton efflux rate, mitochondrial- and glycolysis-associated acidification, after which the acidification rate upon mitochondria and glycolysis inhibition were calculated. All results were normalized using a sulforhodamine B (SRB) assay [75]. Briefly, cells were fixed with 50 μl of 60% trichloroacetic acid (TCA), overnight at 4 °C. After removing the fixing solution and washing with distilled water, fixed cellular proteins were stained with SRB solution (0.05% w/v in 1% acetic acid) for 1 h at 37 °C. Next, unbound dye was removed with 1% acetic acid, and cell bound dye solubilized with 10 mM Tris base solution, pH 10. Colour intensities were measured at 540 nm using a Cytation 3 microplate reader (Biotek, Winooski, VT, USA).
2.12 Alanine aminotransferase activity assay
Alanine aminotransferase (ALT) activity in BPTES-treated cells was determined using a commercial kit (Spinreact) according to the manufacturers’ instructions. ALT catalyses the reversible transfer of an amino group from alanine to α-ketoglutarate forming pyruvate and glutamate. The pyruvate produced is reduced to lactate by LDH together with the oxidation of NADH to NAD+. The rate of decrease in NADH concentration, measured spectrophotometrically, is proportional to the catalytic concentration of ALT present in the sample. The enzymatic activities were calculated by measuring variations in absorbance (340 nm). The activities obtained were calculated as U/μg of protein.
2.13 Oil-red assay
LNCaP (3 × 105 cells/well), DU145 (2 × 105 cells/well) and PC3 (2 × 105 cells/well) cells seeded in 12-well plates and exposed to BPTES (10 μM) for 24 h, were fixed with 4% PFA for 30 min. Next, the cells were washed twice with distilled water and rinsed with 60% isopropanol for 5 min. After an additional wash, the cells were stained with Oil Red O for 20 min. Next, Oil Red O was eluted from the cells with 100% isopropanol under gentle agitation for 5 min. The lipid content was determined by measuring absorbance at 492 nm using a xMark™ Microplate Absorbance Spectrophotometer (Bio-Rad).
2.14 Quantification of free fatty acid
The free fatty acid content in LNCaP, PC3 and DU145 BPTES-treated cells was determined by spectrophotometric analysis using a commercial kit (Sigma-Aldrich, #MAK044) according to the manufacturer’s instructions.
2.15 Statistical analysis
Significant differences among experimental groups were evaluated by unpaired T-test with Welch’s correction or one-way ANOVA followed by Tukey post-hoc test, using GraphPad Prism v6.00 (GraphPad Software, Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant. All experimental data are presented as mean ± standard error of the mean (S.E.M).
3 Results
3.1 CRPC cells are more sensitive to glutamine availability and exhibit a differential expression of ASCT2 and glutaminase compared to LNCaP cells
The expression of the glutamine metabolism regulators ASCT2 and glutaminase (Fig. 1a) in PCa cell models was evaluated using Western blot analysis. We found that the expression of the glutamine transporter ASCT2 was significantly lower in CRPC cells (DU145 and PC3) compared to that in the androgen-sensitive LNCaP cells (0.57 ± 0.07 and 0.61 ± 0.02 fold variation, respectively, Fig. 1b). On the other hand, we found that glutaminase expression (Fig. 1a) was significantly higher in DU145 and PC3 cells compared to that in LNCaP cells (3.66 ± 0.26 and 2.686 ± 0.22, fold variation, respectively, Fig. 1b). Amongst all PCa cell lines tested, DU145 showed the highest expression of glutaminase (Fig. 1b).

ASCT2 and glutaminase expression and sensitivity to glutamine availability in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cell line models. a Schematic representation of glutamine metabolism. Glutamine uptake from the extracellular space occurs through the ASCT2 transporter, whose translocation to the cell membrane via the endoplasmic reticulum depends on post-translational modification, i.e., N-linked glycosylation. At the mitochondria, glutaminase (GLS) converts glutamine into glutamate that enters the tricarboxylic acid (TCA) cycle generating ATP. b ASCT2 and glutaminase expression determined by Western blot analysis after normalization to α-tubulin. Representative blots are shown in the right panel. c Viability of PCa cells under decreasing glutamine concentrations (2, 1, 0.75, 0.5, 0.25, 0 mM). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05; ** p < 0.01; *** p < 0.001 when compared to LNCaP cells and # p < 0.05 when compared to DU145 cells
The viability of LNCaP, DU145 and PC3 cells exposed to decreasing concentrations of glutamine (2, 1, 0.75, 0.5, 0.25 and 0 mM) was evaluated using a MTT assay. We found that glutamine concentrations lower than 0.5 mM significantly decreased the viability of all cell lines tested (Fig. 1c). However, this effect was more pronounced in CRPC cells. The viability of DU145 (0.25 and 0 mM) and PC3 (0 mM) cells was significantly lower than that of LNCaP cells (61.80 ± 3.07% and 50.86 ± 1.16% fold variation, and 59.21 ± 0.28% fold variation, respectively, Fig. 1c).
3.2 DHT stimulates glutamine metabolism in LNCaP cells and rat prostate cells in vivo
Androgens have been shown to play a role as metabolic regulators fuelling PCa cell growth and proliferation [38, 52, 80]. Here, we evaluated the effect of DHT (10 nM) in regulating glutamine consumption (Fig. 2a), as well as ASCT2 (Fig. 2b, d) and glutaminase (Fig. 2c, d) expression in AR-positive and -negative PCa cells. We found that DHT treatment significantly augmented glutamine consumption in the androgen-sensitive LNCaP cells after 24 h of treatment (1.36 ± 0.15 fold-change vs. control, Fig. 2a). Moreover, DHT significantly increased the expression levels of ASCT2 and glutaminase in these cells upon 24 h and 48 h of exposure (ASCT2, 1.68 ± 0.27 and 1.98 ± 0.33 fold-changes vs. control, respectively, Fig. 2b, d; glutaminase, 1.20 ± 0.06 and 1.26 ± 0.12 fold-changes vs. control, respectively, Fig. 2c, d). Overall, no effect was observed on the expression levels of ASCT2 (Fig. 2b, d) and glutaminase (Fig. 2c, d) in the AR-negative CRPC DU145 and PC3 cells in response to DHT.

Effect of DHT (10 nM) on glutamine consumption, ASCT2 and glutaminase expression, and ASCT2 subcellular localization in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells treated for 12, 24 or 48 h. a Glutamine consumption in LNCaP cells determined spectrophotometrically using a commercial kit. b, c Protein expression determined by Western blotting after normalization to β-actin. All results are expressed as fold-change relative to the control untreated group (0 nM DHT, dashed line). d Representative blots. e ASCT2 co-localization with the endoplasmic reticulum marker calnexin in LNCaP cells treated with DHT for 24 h. Co-localization was assessed by immunofluorescence and calculated using the Pearson’s correlation coefficient. f Representative confocal microscopy images showing co-localization of ASCT2 (green) with calnexin (red). Nuclei are stained blue with Hoechst 33342. Negative controls for ASCT2, obtained by omission of the primary antibody, are provided as insert panels (−). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05, ** p < 0.01 when compared to control
Recently, it has been shown that N-linked glycosylation serves as an important post-translational modification required for ASCT2 trafficking from the endoplasmic reticulum to the cell membrane (Fig. 1a) [19]. It has also been shown that androgens can upregulate global protein glycosylation in PCa cells [54], and alter the distribution of several molecular targets across cell compartments [40, 49, 69]. Therefore, we next investigated whether DHT stimulation may affect ASCT2 subcellular localization. For this purpose, ASCT2 co-localization with calnexin was analysed using confocal microscopy (Fig. 2e, f). Calnexin is a membrane protein of the endoplasmic reticulum that functions as a molecular chaperone responsible for glycoprotein folding and quality control [85]. We found that DHT treatment increased the co-localization of ASCT2 with the endoplasmic reticulum protein calnexin by approximately 50% (1.47 ± 0.14 fold-change vs. control, Fig. 2e), as determined by immunofluorescence staining (Fig. 2f) and analysed by the Pearson’s correlation coefficient (Fig. 2e).
To assess whether the increased expression levels of the glutamine regulators ASCT2 and glutaminase in response to DHT in LNCaP cells (Fig. 2) could be recapitulated in vivo, animal stimulation experiments were performed. To this end, orchidectomised rats were treated with DHT (ORCHX + DHT, 500 μg/kg/day) for 5 days. We found that both the ASCT2 and glutaminase expression levels were significantly augmented in the ORCHX + DHT group compared to the ORCHX + Vehicle and/or intact groups (Fig. 3). The ASCT2 and glutaminase expression levels changed 2.06 ± 0.24 and 1.61 ± 0.11 fold relative to those in intact animals, respectively, Fig. 3). No effect was perceived on the protein expression levels of ASCT2 and glutaminase upon castration only (Fig. 3).

Effect of DHT (500 μg/kg/day) on ASCT2 and glutaminase expression levels in rat dorsolateral prostate cells. Three months old orchidectomized (ORCHX) animals received intraperitoneal injections of vehicle (ORCHX + vehicle) or DHT (ORCHX + DHT) for 5 consecutive days. The intact group includes animals that did not undergo surgery and received vehicle only. Protein expression was determined by Western blotting after normalization to β-actin. All results are expressed as fold-change relative to the control intact group. Error bars indicate mean ± S.E.M (n = 5); * p < 0.05 when compared to intact group; # p < 0.05; when compared to ORCHX + vehicle group. Representative blots are shown in the right panel
3.3 Inhibition of glutamine metabolism decreases PCa cell viability and migration, and increases caspase-3-like activity
The viability of LNCaP, DU145 and PC3 cells was determined by MTT assay after treatment with BPTES, a specific glutaminase inhibitor, which has been used to inhibit glutamine metabolism [66]. All BPTES concentrations tested (1–50 μM) significantly decreased the viability of DU145 and PC3 cells (Fig. 4a), though not always in a concentration-dependent manner. BPTES also decreased the viability of LNCaP cells. Here, however, the effect was seen only at the highest (25 μM and 50 μM) concentrations (78 ± 3% and 73 ± 2% cell viability vs. control, respectively, Fig. 4a). Moreover, 1 μM and 2 μM BPTES increased the viability of LNCaP cells (141 ± 9% and 127 ± 8% compared to the control, Fig. 4a). Accordingly, calculated IC50 concentrations for the CRPC and LNCaP cells were as follows, PC3 (3.00 to 4.42 μM, confidence interval 95%) < DU145 (4.63 to 6.97 μM, confidence interval 95%) < LNCaP (above the concentration range).

Effect of BPTES (1–50 μM) on the viability, migration, caspase-3-like activity and p21 and c-Myc protein expression levels in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells treated for 24 h. a LNCaP, DU145 and PC3 cell viability determined by MTT assay (% of control). b PCa cell migration determined by a transwell assay in uncoated chambers. The upper chamber contained serum-free medium and cells in the presence or absence of BPTES (10 μM). FBS supplemented medium in the lower chamber was used as chemoattractant. Data are expressed as the mean number of migrating cells per 20x magnification field (10 fields were assessed for each experimental condition). c Caspase-3-like activity in 10 μM BPTES-treated cells measured spectrophotometrically based on the release of pNA chromophore. d Protein expression upon BPTES treatment (10 μM) determined by Western blot analysis after normalization to β-actin. e Representative blots. Results are expressed as fold-change relative to the control untreated group (0 nM BPTES, dashed line). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05; ** p < 0.01; *** p < 0.001 when compared to the control
The effects of BPTES on the migration and apoptosis of PCa cells were assessed using a single concentration of 10 μM, according to previously reported studies in other cancer cell types [15, 86]. We found that BPTES supplementation decreased the migration capacity of LNCaP, DU145 and PC3 cells in trans-well assays (0.70 ± 0.04, 0.75 ± 0.03 and 0.61 ± 0.05 fold-changes vs. control, respectively, Fig. 4b).
The activity-like level of the executioner caspase-3, as an end-point indicator of apoptosis, was measured in the cell lines after treatment with BPTES. We found that caspase-3-like activities were significantly increased in DU145 and PC3 cells (1.28 ± 0.06 and 1.20 ± 0.06 fold-change vs. control, respectively, Fig. 4c), whereas no differences were detected in LNCaP cells (Fig. 4c).
Subsequent Western blot analysis showed that the alterations observed in PCa cell fate after BPTES treatment were accompanied by alterations in the expression of the cell cycle inhibitor p21 and the oncogene c-Myc (Fig. 4d, e). p21 expression was increased in response to BPTES treatment in all PCa cells tested (2.05 ± 0.40, 1.60 ± 0.06 and 1.52 ± 0.18 fold-changes vs. control, respectively, Fig. 4d). On the other hand, BPTES significantly decreased the expression of c-Myc in LNCaP and PC3 cells (0.59 ± 0.06 and 0.75 ± 0.05 fold-changes vs. control, respectively, Fig. 4d).
3.4 Bicalutamide increases the sensitivity of LNCaP cells to BPTES
Next, we investigated the effect of BPTES on PCa cell viability in the presence of the anti-androgen bicalutamide. For this purpose, LNCaP, DU145 and PC3 cells were treated with BPTES (10 μM) and a concentration range of bicalutamide (10–40 μM). We found that 10 μM BPTES plus 40 μM bicalutamide had an additive effect on suppressing the viability of the androgen-sensitive LNCaP cells (54 ± 8% of control vs 78 ± 4% for BPTES alone, Fig. 5a). No additive effect was perceived for BPTES and bicalutamide on CRPC cells (Fig. 5b, c). On the other hand, we found that DHT supplementation attenuated the effectiveness of BPTES in decreasing LNCaP cell viability (95.61 ± 2.08 vs 75.75 ± 2.90%, Fig. 5d).

Effect of BPTES (10 μM) on the viability of androgen-sensitive (LNCaP) and CRCP (DU145 and PC3) cells depending on bicalutamide (Bica, 10–40, μM) and DHT (10 nM). Cells were cultured for 24 h and viabilities were determined by MTT assay. a, b, c Results expressed as % of control. Error bars indicate mean ± S.E.M (n = 5). * p < 0.05, ** p < 0.01, *** p < 0.001 when compared with the control. $ p < 0.05, $$$ p < 0.001 when compared with bicalutamide (40 μM); ## p < 0.01 when compared with BPTES (10 μM) d Results expressed as % of control. Error bars indicate mean ± S.E.M (n = 5); ** p < 0.01, *** p < 0.001 when compared with control; $ p < 0.05, $$$ p < 0.001 when compared with BPTES (+); ### p < 0.001 when compared with DHT (+) BPTES (+)
3.5 Glycolytic metabolism of PCa cells is affected by inhibition of glutaminase
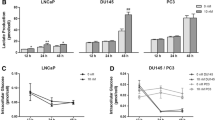
Cancer cells display astonishing metabolic plasticity, reprogramming metabolic routes according to the environmental conditions and nutrients available [13, 68]. It has also been reported that the glycolytic metabolism of PCa cells can be modulated by an increased expression of glutaminase [61]. With this rationale, we next investigated whether glutaminase inhibition resulting from BPTES treatment may disturb the glycolytic metabolism of PCa cells. Glucose consumption and lactate production were determined spectrophotometrically, and the protein levels of key transporters and enzymes involved in glucose metabolism were evaluated by Western blot analysis. We found that BPTES treatment decreased glucose consumption in all PCa cells tested (0.83 ± 0.04, 0.97 ± 0.01 and 0.86 ± 0.02 fold-changes vs. control in LNCaP, DU145 and PC3 cells, respectively, Fig. 6a). This decrease was accompanied by a decreased production of lactate in LNCaP cells (0.83 ± 0.04 fold-change vs. control, Fig. 6b), whereas no significant differences were observed in the other two cell lines (Fig. 6b).

Glucose consumption, lactate production and GLUT1, GLUT2, GLUT3, PFK1, LDH and MCT4 protein levels in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells treated with BPTES (10 μM). a, b Glucose consumption and lactate production determined spectrophotometrically using commercial kits. c Protein expression determined by Western blotting after normalization to β-actin. d Representative blots. The results are expressed as fold-change relative to the control untreated group (0 nM BPTES, dashed line). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05; ** p < 0.01, when compared with control
Glucose consumption is mediated by the uptake of glucose through glucose transporters (GLUTs), i.e., GLUT1, GLUT2 and GLUT3, which have been detected in PCa cells [14, 79]. Here, we found that BPTES treatment differently affected GLUT expression levels in our PCa cell models (Fig. 6c, d). GLUT1 expression was significantly decreased after BPTES treatment in PC3 cells (0.65 ± 0.07 fold-change vs. control, Fig. 6c) and diminished expression of GLUT2 upon BPTES treatment was observed in LNCaP cells (0.59 ± 0.05 fold-change vs. control, Fig. 6c). Concerning GLUT3, BPTES exposure diminished its expression only in DU145 cells (0.73 ± 0.04 fold-change vs. control, Fig. 6c).
Considered to be one of the rate-limiting steps in glycolysis, the conversion of fructose 6-phosphate into fructose 1,6-bisphosphate by PFK1 is a crucial step in the glycolytic flux. We found that BPTES treatment significantly decreased PFK1 expression in all PCa cell lines tested (0.66 ± 0.07, 0.69 ± 0.07 and 0.81 ± 0.05 fold-changes vs. control in PC3, DU145 and LNCaP, respectively, Fig. 6c). Also, a decreased expression of LDH, the enzyme that converts the end-product of glycolysis pyruvate into lactate, was observed in all PCa cell lines tested in response to BPTES (0.68 ± 0.08, 0.73 ± 0.07 and 0.87 ± 0.03 fold-changes vs. control in PC3, DU145 and LNCaP, respectively, Fig. 6c). The lactate produced is exported to the extracellular space through specific members of the monocarboxylate transporter (MCT) family, i.e., MCT4, which has been associated with PCa progression and poor prognosis [3, 17, 79]. Here, we found that MCT4 expression was significantly decreased upon BPTES treatment in LNCaP cells (0.59 ± 0.07 fold-change vs. control, Fig. 6c).
A Seahorse XF Glycolytic Rate Assay was used to investigate the effect of BPTES on glycolytic metabolism in PCa cells. Specifically, the contribution of glycolysis to extracellular acidification before and after mitochondrial inhibition was evaluated. Pyruvate from glycolysis can be converted to lactate in the cytosol or, alternatively, to CO2 and H2O in the mitochondria. Both pathways result in acidification of extracellular media by the extrusion of protons or by the reaction of CO2 with water forming carbonic acid, which also produces protons (Fig. 7a). The basal proton efflux rate (PER) represents the contribution of both pathways to the acidification of cell culture medium under normal culture conditions (Fig. 7b). We found that the basal PER was significantly decreased after BPTES treatment in LNCaP and PC3 cells (0.60 ± 0.04 and 0.83 ± 0.05 fold-changes vs. control, respectively, Fig. 7c), whereas it remained unaltered in DU145 cells (Fig. 7c). Subtracting the contribution from the effect of CO2-dependent acidification (mitochondrial-associated acidification) provides the contribution of glycolysis (Glyco PER, Fig. 7b). We found that BPTES treatment decreased Glyco PER only in LNCaP cells (0.64 ± 0.05 fold-change vs. control, Fig. 7c), which was correlated with the observed reduction in lactate extrusion rate (Fig. 6b). Compensatory Glycolysis (Fig. 7b) indicates the maximum cell capability to use glycolysis after mitochondrial inhibition. We found that adding rotenone and antimycin A, inhibitors of mitochondrial electron transport chain Complex I and III, respectively, upon BPTES-treatment resulted in a significant diminution of Compensatory Glycolysis in all PCa cell lines tested (0.70 ± 0.04, 0.83 ± 0.04 and 0.71 ± 0.04 fold-changes vs. control in DU145, LNCaP and PC3 cells, respectively, Fig. 7c). Post-2-DG acidification (Fig. 7b), obtained after inhibition of glycolysis by 2-DG injection, induced a differential response in LNCaP and CRPC cells. BPTES treatment significantly increased Post-2-DG acidification in LNCaP cells (1.70 ± 0.28 fold-change vs. control, Fig. 7c), whereas the opposite effect was observed in DU145 and PC3 cells (0.80 ± 0.06 and 0.63 ± 0.06 fold-changes vs. control, respectively, Fig. 7c).

Effect of BPTES (10 μM) on glycolysis and extracellular acidification in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells. a Glycolysis and mitochondrial respiration as cell energy sources. Glucose is transformed into lactate during glycolysis, with the resulting protons from NADH conversion to NAD+ being extruded into the extracellular space. Mitochondrial activity (TCA cycle and oxidative phosphorylation) produces CO2 and H2O and generates H+, which contributes to the acidification of the surrounding environment. b Schematic panel illustrating the experimental strategy. Proton efflux rate (PER) was obtained using the Seahorse XF96 Glycolytic Rate Assay. Basal PER represents the initial total PER. The use of rotenone and antimycin A (0.5 μM), inhibitors of mitochondrial complexes I and III, respectively, enabled calculation of the mitochondrial-associated acidification (Mito PER). The Glycolytic PER (Glyco PER) resulted from subtracting Mito PER from Basal PER results. Inhibition of mitochondria drives compensatory changes to use glycolysis to meet energy demands (Compensatory Glycolysis). Glycolysis was inhibited using the glucose analogue 2-deoxy-D-glucose (2-DG, 50 mM, Post-2-DG acidification). c Basal PER, Glycolytic PER, Compensatory Glycolysis and Post 2-DG Acidification in LNCaP, DU145 and PC3 BPTES-treated cells. Each experimental result was normalized to cell mass determined by SRB assay. The results are expressed as fold-change relative to the control untreated group (0 nM BPTES, dashed line). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05; ** p < 0.01, *** p < 0.001 when compared with control
Finally, extracellular acidification can occur through glucose- and glutamine-independent pathways, i.e., through the activity of aminotransferases. The aminotransferase ALT catalyzes a reversible conversion of alanine into glutamate and pyruvate [46]. Here, we found that BPTES treatment significantly increased ALT activity in LNCaP cells (2.60 ± 0.63 fold-change vs. control, Fig. 8), whereas a diminution was observed in PC3 cells (0.44 ± 0.05 fold-change vs. control, Fig. 8).

Effect of BPTES (10 μM) on ALT activity in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells. All results are expressed as fold-change relative to the untreated control group (0 nM BPTES, dashed line). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05; ** p < 0.01 when compared with control
3.6 Glutaminase inhibition affects lipid metabolism
Glutamine and glutamate are the main suppliers of the anaplerotic reaction that produces citrate, which in turn is the principal source for lipid synthesis during cancer cell growth [10, 23]. A relationship between glutaminolysis and fatty acid metabolism has been reported [2, 10, 82]. Based on this information, we next evaluated the expression of FASN, a crucial enzyme in fatty acid synthesis, and CPT1A, a rate-limiting component in the carnitine-dependent transport of fatty acids across the inner mitochondrial membrane, upon glutaminase inhibition (Fig. 9a). We found that androgen-sensitive and CPRC cells exhibited different responses to BPTES (Fig. 9). FASN expression was significantly decreased in BPTES treated LNCaP cells (0.48 ± 0.06 fold-change vs. control, Fig. 9a), but increased in BPTES treated PC3 cells (1.77 ± 0.24 fold-change vs. control, Fig. 9a). Similarly, CPT1A expression was decreased in LNCaP cells (0.73 ± 0.077 fold-change vs. control, Fig. 9a), but increased in DU145 and PC3 cells (2.9 ± 0.46 and 2.10 ± 0.14 fold-changes vs. control, respectively, Fig. 9a) upon exposure to BPTES. Next, we evaluated the free fatty acid content in PCa cells upon BPTES treatment, and found that it was decreased in PC3 cells (0.92 ± 0.013 fold-change vs. control, Fig. 9b). Acumulation of lipids in cytosolic lipid droplets is a feature of cancer cells involving increased lipid uptake, de novo lipid synthesis and lipid remodelling, being an additional source of fatty acids from lipolysis [21, 59]. We found that inhibition of glutaminase by BPTES decreased the lipid content of LNCaP cells (0.86 ± 0.019 fold-change vs. control, respectively, Fig. 9c).

Effect of BPTES (10 μM) on the expression of FASN and CPT1A, and free fatty acid and lipid content in androgen-sensitive (LNCaP) and CRPC (DU145 and PC3) cells. a Protein expression determined by Western blotting after normalization to β-actin. b Free fatty acid content determined spectrophotometrically using a commercial kit. c Lipid content quantified by lipid droplet staining and elution using an Oil Red-O assay. All results are expressed as fold-change relative to the control untreated group (0 nM BPTES, dashed line). Error bars indicate mean ± S.E.M (n = 5); * p < 0.05; ** p < 0.01 when compared with control. Representative blots are shown in the right panel
4 Discussion
Our data revealed the importance of glutaminolysis in maintaining the survival and migration abilities of androgen-sensitive and AR-negative CRPC cells. In addition, we found that glutaminase inhibition may have an impact on other metabolic pathways, i.e., those involved in glycolysis and lipid metabolism.
As a starting point, we found that the different PCa cell line models used, i.e., LNCaP (androgen-sensitive) cells and CRPC DU145 and PC3 cells, displayed distinct expression levels of the key regulators of glutamine metabolism, the glutamine transporter ASCT2 and glutaminase. Here, we focused on ASTC2, as it is the receptor that has been shown to be responsible for glutamine uptake and tumor growth [20, 87]. CRPC cells displayed lower basal ASCT2 expression levels compared to LNCaP cells. Previously, ASCT2 expression has been shown to be increased in primary PCa samples compared to non-cancer tissues [81]. On the other hand, it has been found that patients undergoing anti-androgen therapy displayed a significant decrease in prostate ASCT2 expression compared to untreated individuals [81], which is concordant with the expression pattern we observed in the androgen-sensitive and CRPC cell line models.
In contrast, glutaminase expression was found to be significantly higher in the more aggressive CRPC cells relative to LNCaP cells. Previous patient data revealed overexpression of glutaminase in PCa tissues compared to benign prostate hyperplasia or non-cancer tissues, which positively correlated with Gleason scores [61, 94], thereby also linking increased glutaminase expression with the aggressiveness of the disease. A higher expression of glutaminase in PCa cells relative to the normal prostatic epithelial cell line RWPE-1 has also previously been reported [94]. Our present study is the first showing differential glutaminase expression between castration-resistant (AR-negative) and androgen-sensitive PCa cell line models, which is in accordance with the patient findings. Our data also revealed that restricting glutamine availability suppresses the viability of PCa cells, with a relatively higher impact on CRPC cells. Under low concentrations or glutamine deprivation, the viability of CRPC cells was significantly lower than that of androgen-sensitive LNCaP cells, which indicates that CRPC cells are more glutaminolytic and dependent on the utilization of glutamine to survive.
Androgens are widely recognized as principal hormones responsible for maintaining prostate cell proliferation and survival [6, 88]. These hormones have also been pointed out as important regulators of prostate cell metabolism, including the modulation of lipid metabolism and glycolysis [6, 38, 52, 80, 88]. However, the action of androgens in the regulation of glutamine metabolism is less well known. Here, we show that DHT treatment stimulates glutaminolysis in LNCaP cells, which was underscored by an enhanced glutamine consumption and an increased expression of both the amino acid transporter ASCT2 and glutaminase. Moreover, these in vitro results were mirrored in vivo in the rat prostate, where DHT treatment upregulated the expression levels of ASCT2 and glutaminase. Intracellular glutamine can be converted to glutamate through glutaminase, and the observed increased expression of this enzyme is in line with the observed augmentation in glutamine uptake in response to DHT. These findings corroborate a previous study showing that androgens may regulate glutaminase expression in other androgen-sensitive (VCaP) PCa cells [84].
ASCT2 is the major molecular player in mediating glutamine uptake and, thus, its increased expression levels clearly support the enhanced glucose consumption observed in LNCaP cells. Indeed, other studies have shown upregulation of ASCT2 expression in LNCaP and VCaP cells in response to androgens [60, 84], which was also accompanied by an increased uptake of glutamine [84]. Human ASCT2 contains two N-glycosylation sites (N163 and N212) that, although not altering the activity of the protein, have been shown to increase its stability, and to be responsible for intracellular trafficking from the endoplasmic reticulum to the plasma membrane [19]. Aberrant glycosylation has been identified as a typical event in PCa and to be regulated by androgens [43, 54,55,56]. These hormones are also capable of altering the subcellular localization of several molecular targets [40, 49, 69]. Therefore, we sought to investigate whether exposure to DHT affects the intracellular distribution of ASCT2. Indeed, we found that DHT treatment significantly increased co-localization of ASCT2 with the endoplasmic reticulum chaperone calnexin, which coordinates N-linked protein glycosylation, glycoprotein folding and quality control [36, 67, 85]. The endoplasmic reticulum is the major organelle involved in protein glycosylation and a central player in the distribution of proteins to the cell membrane through the endoplasmic reticulum-Golgi apparatus pathway. Thus, our findings support the role of androgens driving the trafficking of ASCT2 from the endoplasmic reticulum to the plasma membrane. Although future studies are needed to clarify the role of androgens in regulating glycosylation and intracellular trafficking of ASCT2, here we show co-localization of ASCT2 with the endoplasmic reticulum. Moreover, the increased co-localization of ASCT2 with calnexin was concomitant with the augmented glutamine consumption in LNCaP cells, supporting an increased density of the transporter molecules at the cell membrane. Together, these observations substantiate the role of androgens as central regulators of glutamine metabolism in PCa.
Considering the androgenic regulation of glutamine metabolism and the distinct sensitivity of CRPC and LNCaP cells to glutamine availability, the cell fate and metabolic response upon inhibition of glutaminolysis were investigated. BPTES, a recognized inhibitor of glutaminase [66], decreased the viability of all PCa cell lines studied. However, CRPC and androgen-sensitive cells displayed distinct responses to BPTES, with low concentrations of glutaminase inhibitor (1–10 μM) only effectively suppressing the viability of DU145 and PC3 cells. This is further supported by the finding that CRPC cells were more sensitive to a limited availability of glutamine. Interestingly, DU145 and PC3 cells exhibited higher glutaminase basal expression levels compared to LNCaP cells, but this may not be the only reason underlying the higher sensitivity of CRPC cells to BPTES. Inhibition of glutaminolysis by enzyme inhibitors, such as BPTES or CB-839, or silencing glutaminase expression has also been shown to decrease the proliferation of several cancer cell types, including PCa cells [9, 34, 61, 77]. Moreover, our present findings underscore a previous report showing that cancer cells that more actively use glutamine are more sensitive to inhibition of this metabolic pathway [91]. Taken together, the results obtained indicate that CRPC cells are more dependent on the utilization of glutamine as energy source to support their growth than androgen-sensitive LNCaP cells.
In line with the cell viability results, we found that glutaminase inhibition with BPTES also induced apoptosis of CRPC DU145 and PC3 cells, as indicated by an augmented caspase-3-like activity. Caspase-3 is the executioner caspase at the convergence of intrinsic and extrinsic apoptotic pathways. Thus, activation of precursor caspase-3 is a recognized indicator of apoptosis [41, 58]. Nevertheless, we found that BTPES-modulated cell behavior was associated with altered expression of cell cycle and oncogenic pathway regulators in all PCa cell lines tested. We found, for example, that the expression of the potent cyclin-dependent kinase inhibitor p21, a cell cycle inhibitor functioning as tumor suppressor and activator of apoptosis by cleavage of precursor caspase-3 [31, 93], was significantly increased in LNCaP, DU145 and PC3 cells. Although there are no reports on the inhibition of glutaminase by compound 968 in PCa, in ovarian cancer cells this compound has also been found to increase p21 expression, concomitantly with reduced cyclin-dependent kinase 4 and cyclin D levels [90].
c-Myc is one of the oncogenes known to be overexpressed in PCa, being important for cancer progression and establishment of the CRPC phenotype [5, 7, 11]. Moreover, c-Myc has been shown to be involved in the metabolic reprogramming of PCa cells, specifically promoting the expression of glutaminase [30, 63]. Noteworthy, we found that c-Myc expression decreased in LNCaP and PC3 cells after treatment with BPTES. Although mechanisms involved in the interplay between c-Myc expression and glutaminase inhibition in PCa cells still need to be clarified, BPTES inhibition in renal cell carcinoma has been found to halt renal cancer progression driven by c-Myc [70].
Besides its effects on cell viability and caspase-3-like activity, we found that inhibition of glutaminase also has the potential to decrease the migratory capabilities of PCa cells, effects that were transversal to all cell lines. These findings are in agreement with similar reports in oral, lung and breast cancer cells, where glutaminase activity was shown to be related to the promotion of cancer cell migration [37, 65, 83]. Overall, our data revealed an effectiveness of the glutaminase inhibitor BPTES in decreasing the viability, inducing the apoptosis and suppressing the migration of CRPC cells, with only moderate effects in hormone-sensitive LNCaP cells.
ADT remains the mainstream therapy for hormone-sensitive primary and metastatic PCa [62, 76]. However, tumours that initially respond to therapy inevitably acquire resistance to therapy and progress to more aggressive stages, i.e., the castration resistant form of the disease [62, 76]. The development of CRPC is a complex process relying on abnormal activation of the AR signalling axis, driven by e.g. the occurrence of genetic variants, positive and negative AR regulators, altered post-translational AR modifications and ligand-independent AR activation [27, 28]. Recently, it has been shown that metabolic reprogramming underlies the establishment of the androgen independent phenotype and the occurrence of treatment resistance, which includes changes in glucose, lipid and glutamine metabolism [38, 91, 92]. The metabolic peculiarities of PCa cells and its potential therapeutic advantages have been gaining interest [13]. In fact, in vitro and in vivo evidence shows that combination of ADT with, for example, lipid or cholesterol metabolism blockage may have a positive effect on suppressing cancer cell growth and overcoming ADT resistance [29, 47, 92]. Here, we found that the concomitant administration of BPTES and bicalutamide had an additive effect on diminishing the viability of androgen-sensitive LNCaP cells, whereas DHT suppressed the effect of inhibiting glutaminase. These findings open up new perspectives for the development of co-treatment approaches using anti-androgens and metabolic inhibitors, i.e., glutaminase inhibitors.
Cancer cell metabolism comprises a complex mixture of diverse and intricate processes that depend on a variety of nutrients fuelling the TCA cycle. Previous reports have shown that a relationship exists between glucose and glutamine handling, and that glutamine availability can change glucose uptake and metabolism [22, 61]. Glutamine is converted to glutamate by glutaminase and subsequently to α-ketoglutarate. It has been shown that α-ketoglutarate can directly regulate glucose uptake [24, 72]. Moreover, glutamine-derived glutamate and α-ketoglutarate have been linked to the electron transfer system in the malate-aspartate shuttle between mitochondria and the cytosol, which allows the regeneration of NAD+ for glycolysis [48]. Suppression of glutamate production can impair this shuttle, thereby affecting glycolysis and oxygen consumption. Also, metabolic crosstalk exists between glucose, glutamine and their intermediate metabolites. Pyruvate, the end product of glycolysis, can also be obtained from glutamine by ALT, which catalyses the reversible transfer of an amino group from alanine to α-ketoglutarate forming pyruvate and glutamate [1]. Here, we found that suppression of glutaminase activity by BPTES significantly reduced glucose consumption in all PCa cells tested. Similar effects have been described in PCa PC3 cells upon siRNA-mediated glutaminase silencing or BPTES administration, resulting in a decrease in glucose uptake [61].
Glucose consumption starts with its uptake from the extracellular medium through GLUT family members, i.e., GLUT1, GLUT2 and GLUT3, in PCa cells [14, 79]. We found that BPTES treatment decreased glucose consumption in all PCa cell line models tested, which was supported by a decreased expression of GLUTs. A differential response was observed for different GLUT family members, i.e., treatment with BPTES decreased GLUT1, GLUT2 or GLUT3 levels in PC3, LNCaP and DU145 cells, respectively. Glucose uptake by distinct GLUTs in different PCa cells was also observed in another study [12].
A crucial step in the glycolytic process is the irreversible conversion of fructose 6-phosphate to fructose 1,6-bisphosphate, a reaction catalysed by PFK1. We found that BPTES-treated PCa cells exhibited a diminished expression of PFK1, which indicates that besides a reduced glucose consumption, the glycolytic flux rate is decreased upon glutaminase inhibition. Although a possible correlation between PFK1 activity and glutamine metabolism in PCa cells is currently unknown, in Hela cells both glucose and glutamine were found to be required for cell cycle progression, which was also related with a decreased activity of APC/C-Cdh1 ubiquitin ligase leading to accumulation of the glutaminase and glycolysis-promoting enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase, isoform 3 [18].
The last step in glycolytic metabolism is the conversion of pyruvate to lactate through LDH, followed by the export of lactate to the extracellular space. The reduced glucose consumption observed in BPTES-treated LNCaP cells was accompanied by a decreased export of lactate, implicated by a decreased expression of LDH and MCT4. Although information in PCa is very limited or non-existent, inhibition of glutaminase by compound 968 in ovarian cancer cells also resulted in a decreased lactate export [89]. Similar findings were found in colorectal cancer cells, with glutaminase inhibition suppressing glucose uptake and lactate export [73].
We found that BPTES treatment also decreased the basal efflux of protons (Basal PER) in LNCaP and PC3 cells, which in the case of LNCaP cells seems to result from a direct contribution of glycolysis (Glyco PER). The Glyco PER parameter is directly correlated with the export of lactate, and strictly follows the decreased lactate production observed in LNCaP cells after BPTES treatment. Interestingly, when oxidative phosphorylation was inhibited none of the PCa cell lines tested had the capability to compensate this phenomenon using glycolysis (Compensatory Glycolysis). These results implicated the relevance of glutaminase in influencing the glycolytic pathway and identified glutaminase as a metabolic checkpoint in the interplay between glutamine and glucose metabolism, with impact on cell survival, proliferation and migration.
After inhibiting glutaminolysis and oxidative phosphorylation, the addition of 2-DG halted the glycolytic pathway, disabling PCa cells to use any of these metabolic pathways. CRPC cells treated with BPTES exhibited lower levels of acidification attributed to other sources than glycolysis or mitochondrial TCA activity. Interestingly, the relatively BPTES-resistant LNCaP cells exhibited increased levels of extracellular acidification after inhibition of glutaminolysis, mitochondrial activity and glycolysis. This indicates that these cells are capable of using diverse and alternative energy sources. Therefore, we evaluated the activity of ALT in response to glutaminase inhibition, as this enzyme is placed in the interplay between glycolysis and glutaminolysis by its capability to generate both pyruvate and glutamate. Interestingly, we found that BPTES-treated LNCaP cells exhibited increased ALT activity, whereas a decreased activity was observed in PC3 cells. Similar findings have been described for HeLa cells, with ALT expression being induced by glutamine deprivation or glutaminase inhibition, which was shown to sustain the TCA cycle by the conversion of glutamate to α-ketoglutarate [46]. A compensatory response of cancer cells upon inhibition of glutaminolysis was also observed in pancreatic cancer cells, which displayed increased ALT and glutamate levels and no decreased α-ketoglutarate levels, indicating reactivation of alternative carbon sources [8]. Therefore, it is possible that the source for the extracellular acidification in LNCaP cells after inhibition of glutaminolysis, mitochondrial activity and glycolysis, may result from reactivation of alternative carbon sources likely by ALT activity.
Cancer cell growth is intimately associated with lipid synthesis, which is continuously sustained by the supply of citrate. Several pieces of evidence indicate that cancer cells can generate citrate for fatty acid synthesis from the reductive metabolism of glutamine [10, 23]. In addition, it has been shown that glutamine contributes 25% of lipids synthesized by cancer cells [53]. CRPC cells seem to have the capacity to augment fatty acid synthesis and lipid oxidation in response to glutaminase inhibition, as indicated by an increased expression of FASN and CPT1A, respectively. Accelerated lipid catabolism under inhibited glutaminolysis has also been described in breast cancer cells and to be related with activation of survival mechanisms [35]. Moreover, a previous study reported that inhibition of ASCT2 decreased fatty acid synthesis in LNCaP cells, but not in PC3 cells [81]. The decreased content of fatty acids observed in BPTES-treated PC3 cells, together with an increased FASN and CPT1A expression, suggests that CRPC cells are extensively using lipids as their energy source, with fatty acid β-oxidation superimposing to de novo lipid synthesis. A distinct behavior of LNCaP cells in handling lipids was observed when glutaminolysis was inhibited. We found that decreased FASN and CPT1A expression after BPTES treatment, suggesting diminished de novo lipid synthesis and reduced oxidation, resulted in a balanced content of free fatty acids, though the overall lipid content was decreased.
Together, the here obtained results indicate a pivotal role of glutamine in shaping PCa cell metabolism, i.e., glycolysis and lipid handling. We found that, despite this adaptation, inhibition of glutaminolysis with BPTES significantly altered PCa cell fate. Our findings also indicate that it is crucial to deeply understand the metabolic responses of cancer cells when specific metabolic pathways are targeted for therapy. Future studies should address the adaptive metabolic responses of PCa cells to different metabolic inhibitors to be tested (pre)clinically.
In conclusion, we found that androgens potentiated glutamine metabolism in AR-positive PCa cells, whereas the inhibition of glutaminase activity decreased cell viability and migration, and increased apoptosis, particularly in CRPC cells, which showed to be more glutaminolytic (Fig. 10). These outcomes appear exciting considering targeting glutaminase for CRPC therapy. Using molecular and metabolic extracellular flux analyses, we also found that suppression of glutaminolysis in PCa cells has impact on glycolysis and lipid metabolism (Fig. 10). Our findings highlight the importance of fully understanding cancer metabolic remodelling whenever specific metabolic routes are inhibited with therapeutic purposes.

Glutaminolysis is an essential metabolic route in androgen-sensitive (LNCaP) and CRPC cells. DHT stimulates glutamine uptake and the expression of ASCT2 and glutaminase (GLS) in LNCaP cells. DHT also triggers ASCT2 localization from the endoplasmic reticulum to the cell membrane. GLS inhibition suppresses glycolysis and the expression of FASN (catalyses the conversion of malonyl CoA to fatty acids (FA)) and CPT1A in LNCaP cells. In contrast, FASN and CPT1A expression levels are increased in CRPC cells. Augmented activity of ALT was found in LNCaP cells upon blockage of glutaminolysis. CRPC cells were more glutaminolytic and GLS inhibition decreased their viability and migration, whereas augmenting apoptosis
References
B.J. Altman, Z.E. Stine, C.V. Dang, From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 16, 619–634 (2016)
D. Anastasiou, L.C. Cantley, Breathless cancer cells get fat on glutamine. Cell. Res. 22, 443–446 (2012)
S. Andersen, O. Solstad, L. Moi, T. Donnem, M. Eilertsen, Y. Nordby, N. Ness, E. Richardsen, L.T. Busund, R.M. Bremnes, Organized metabolic crime in prostate cancer: The coexpression of MCT1 in tumor and MCT4 in stroma is an independent prognosticator for biochemical failure. Urol. Oncol. 33, 338.e339–338.e317 (2015)
D.A. Bader, S.M. Hartig, V. Putluri, C. Foley, M.P. Hamilton, E.A. Smith, P.K. Saha, A. Panigrahi, C. Walker, L. Zong, H. Martini-Stoica, R. Chen, K. Rajapakshe, C. Coarfa, A. Sreekumar, N. Mitsiades, J.A. Bankson, M.M. Ittmann, B.W. O’Malley, N. Putluri, S.E. McGuire, Mitochondrial pyruvate import is a metabolic vulnerability in androgen receptor-driven prostate cancer. Nat. Metab. 1, 70–85 (2019)
S. Bai, S. Cao, L. Jin, M. Kobelski, B. Schouest, X. Wang, N. Ungerleider, M. Baddoo, W. Zhang, E. Corey, R.L. Vessella, X. Dong, K. Zhang, X. Yu, E.K. Flemington, Y. Dong, A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 38, 4977–4989 (2019)
P.P. Banerjee, S. Banerjee, T.R. Brown, B.R. Zirkin, Androgen action in prostate function and disease. Am. J. Clin. Exp. Urol. 6, 62–77 (2018)
S.J. Barfeld, A. Urbanucci, H.M. Itkonen, L. Fazli, J.L. Hicks, B. Thiede, P.S. Rennie, S. Yegnasubramanian, A.M. DeMarzo, I.G. Mills, c-Myc antagonises the transcriptional activity of the androgen receptor in prostate cancer affecting key gene networks. EBioMedicine 18, 83–93 (2017)
D.E. Biancur, J.A. Paulo, B. Malachowska, M. Quiles Del Rey, C.M. Sousa, X. Wang, A.S.W. Sohn, G.C. Chu, S.P. Gygi, J.W. Harper, W. Fendler, J.D. Mancias, A.C. Kimmelman, Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 8, 15965 (2017)
G. Boysen, A. Jamshidi-Parsian, M.A. Davis, E.R. Siegel, C.M. Simecka, R.A. Kore, R.P.M. Dings, R.J. Griffin, Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int. J. Radiat. Biol. 95, 436–442 (2019)
S.A. Brose, A.L. Marquardt, M.Y. Golovko, Fatty acid biosynthesis from glutamate and glutamine is specifically induced in neuronal cells under hypoxia. J. Neurochem. 129, 400–412 (2014)
R. Camarda, J. Williams, A. Goga, In vivo reprogramming of cancer metabolism by MYC. Front. Cell. Dev. Biol. 5, 35 (2017)
H.J. Cardoso, C.V. Vaz, T.M.A. Carvalho, M.I. Figueira, S. Socorro, Tyrosine kinase inhibitor imatinib modulates the viability and apoptosis of castrate-resistant prostate cancer cells dependently on the glycolytic environment. Life Sci. 218, 274–283 (2019)
T.M. Carvalho, H.J. Cardoso, M.I. Figueira, C.V. Vaz, S. Socorro, The peculiarities of cancer cell metabolism: A route to metastasization and a target for therapy. Eur. J. Med. Chem. 171, 343–363 (2019)
J.D. Chandler, E.D. Williams, J.L. Slavin, J.D. Best, S. Rogers, Expression and localization of GLUT1 and GLUT12 in prostate carcinoma. Cancer 97, 2035–2042 (2003)
L. Chen, H. Cui, J. Fang, H. Deng, P. Kuang, H. Guo, X. Wang, L. Zhao, Glutamine deprivation plus BPTES alters etoposide- and cisplatin-induced apoptosis in triple negative breast cancer cells. Oncotarget 7, 54691–54701 (2016)
Y.-K. Choi, K.-G. Park, Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 26, 19–28 (2018)
S.Y. Choi, H. Xue, R. Wu, L. Fazli, D. Lin, C.C. Collins, M.E. Gleave, P.W. Gout, Y. Wang, The MCT4 gene: A novel, potential target for therapy of advanced prostate cancer. Clin. Cancer Res. 22, 2721–2733 (2016)
S.L. Colombo, M. Palacios-Callender, N. Frakich, S. Carcamo, I. Kovacs, S. Tudzarova, S. Moncada, Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc. Natl. Acad. Sci. U. S. A. 108, 21069–21074 (2011)
L. Console, M. Scalise, Z. Tarmakova, I.R. Coe, C. Indiveri, N-linked glycosylation of human SLC1A5 (ASCT2) transporter is critical for trafficking to membrane. Biochim. Biophys. Acta. 1853, 1636–1645 (2015)
Y. Cormerais, P.A. Massard, M. Vucetic, S. Giuliano, E. Tambutté, J. Durivault, V. Vial, H. Endou, M.F. Wempe, S.K. Parks, J. Pouyssegur, The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 293, 2877–2887 (2018)
A.L.S. Cruz, E.D.A. Barreto, N.P.B. Fazolini, J.P.B. Viola, P.T. Bozza, Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis. 11, 105 (2020)
C. Damiani, R. Colombo, D. Gaglio, F. Mastroianni, D. Pescini, H.V. Westerhoff, G. Mauri, M. Vanoni, L. Alberghina, A metabolic core model elucidates how enhanced utilization of glucose and glutamine, with enhanced glutamine-dependent lactate production, promotes cancer cell growth: The WarburQ effect. PLoS Comput. Biol. 13, e1005758 (2017)
R.J. DeBerardinis, A. Mancuso, E. Daikhin, I. Nissim, M. Yudkoff, S. Wehrli, C.B. Thompson, Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A. 104, 19345–19350 (2007)
C.D. Doucette, D.J. Schwab, N.S. Wingreen, J.D. Rabinowitz, α-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat. Chem. Biol. 7, 894–901 (2011)
J. El-Amm, J.B. Aragon-Ching, The current landscape of treatment in non-metastatic castration-resistant prostate Cancer. Clin. Med. Insights Oncol. 13, 1179554919833927-1179554919833927 (2019)
A. Elgogary, Q. Xu, B. Poore, J. Alt, S.C. Zimmermann, L. Zhao, J. Fu, B. Chen, S. Xia, Y. Liu, M. Neisser, C. Nguyen, R. Lee, J.K. Park, J. Reyes, T. Hartung, C. Rojas, R. Rais, T. Tsukamoto, G.L. Semenza, J. Hanes, B.S. Slusher, A. Le, Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. U.S.A. 113, E5328–E5336 (2016)
A.A. Farooqi, F.H. Sarkar, Overview on the complexity of androgen receptor-targeted therapy for prostate cancer. Cancer Cell. Int. 15, 7 (2015)
Q. Feng, B. He, Androgen receptor signaling in the development of castration-resistant prostate cancer. Front. Oncol. 9 (2019). https://doi.org/10.3389/fonc.2019.00858
T.W. Flaig, M. Salzmann-Sullivan, L.J. Su, Z. Zhang, M. Joshi, M.A. Gijon, J. Kim, J.J. Arcaroli, A. Van Bokhoven, M.S. Lucia, F.G. La Rosa, I.R. Schlaepfer, Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 8, 56051–56065 (2017)
P. Gao, I. Tchernyshyov, T.C. Chang, Y.S. Lee, K. Kita, T. Ochi, K.I. Zeller, A.M. De Marzo, J.E. Van Eyk, J.T. Mendell, C.V. Dang, c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765 (2009)
A.G. Georgakilas, O.A. Martin, W.M. Bonner, p21: A two-faced genome guardian. Trends Mol. Med. 23, 310–319 (2017)
K. Gonthier, R.T.K. Poluri, E. Audet-Walsh, Functional genomic studies reveal the androgen receptor as a master regulator of cellular energy metabolism in prostate cancer. J. Steroid. Biochem. Mol. Biol. 191, 105367 (2019)
S.M. Green, E.A. Mostaghel, P.S. Nelson, Androgen action and metabolism in prostate cancer. Mol. Cell. Endocrinol. 360, 3–13 (2012)
M.I. Gross, S.D. Demo, J.B. Dennison, L. Chen, T. Chernov-Rogan, B. Goyal, J.R. Janes, G.J. Laidig, E.R. Lewis, J. Li, A.L. Mackinnon, F. Parlati, M.L. Rodriguez, P.J. Shwonek, E.B. Sjogren, T.F. Stanton, T. Wang, J. Yang, F. Zhao, M.K. Bennett, Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 13, 890–901 (2014)
A. Halama, M. Kulinski, S.S. Dib, S.B. Zaghlool, K.S. Siveen, A. Iskandarani, J. Zierer, K.S. Prabhu, N.J. Satheesh, A.M. Bhagwat, S. Uddin, G. Kastenmüller, O. Elemento, S.S. Gross, K. Suhre, Accelerated lipid catabolism and autophagy are cancer survival mechanisms under inhibited glutaminolysis. Cancer Lett. 430, 133–147 (2018)
C. Hammond, I. Braakman, A. Helenius, Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. U. S. A. 91, 913–917 (1994)
T. Han, M. Guo, T. Zhang, M. Gan, C. Xie, J.B. Wang, A novel glutaminase inhibitor-968 inhibits the migration and proliferation of non-small cell lung cancer cells by targeting EGFR/ERK signaling pathway. Oncotarget 8, 28063–28073 (2017)
W. Han, S. Gao, D. Barrett, M. Ahmed, D. Han, J.A. Macoska, H.H. He, C. Cai, Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 37, 710–721 (2018)
D. Hanahan, R.A. Weinberg, Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011)
J.J. Hsiao, B.H. Ng, M.M. Smits, J. Wang, R.J. Jasavala, H.D. Martinez, J. Lee, J.J. Alston, H. Misonou, J.S. Trimmer, M.E. Wright, Androgen receptor and chemokine receptors 4 and 7 form a signaling axis to regulate CXCL12-dependent cellular motility. BMC Cancer 15, 204 (2015)
Q. Huang, F. Li, X. Liu, W. Li, W. Shi, F.-F. Liu, B. O'Sullivan, Z. He, Y. Peng, A.-C. Tan, L. Zhou, J. Shen, G. Han, X.-J. Wang, J. Thorburn, A. Thorburn, A. Jimeno, D. Raben, J.S. Bedford, C.-Y. Li, Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 17, 860–866 (2011)
Y. Huang, X. Jiang, X. Liang, G. Jiang, Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 15, 6063–6076 (2018)
H.M. Itkonen, I.G. Mills, N-linked glycosylation supports cross-talk between receptor tyrosine kinases and androgen receptor. PLoS One 8, e65016 (2013)
G. Ji, G. Song, C. Huang, S. He, L. Zhou, Rapidly decreasing level of prostate-specific antigen during initial androgen deprivation therapy is a risk factor for early progression to castration-resistant prostate cancer: A retrospective study. Medicine (Baltimore) 96, e7823 (2017)
M.E. Kaighn, K.S. Narayan, Y. Ohnuki, J.F. Lechner, L.W. Jones, Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investig. Urol. 17, 16–23 (1979)
M. Kim, J. Gwak, S. Hwang, S. Yang, S.M. Jeong, Mitochondrial GPT2 plays a pivotal role in metabolic adaptation to the perturbation of mitochondrial glutamine metabolism. Oncogene 38, 4729–4738 (2019)
Y. Kong, L. Cheng, F. Mao, Z. Zhang, Y. Zhang, E. Farah, J. Bosler, Y. Bai, N. Ahmad, S. Kuang, L. Li, X. Liu, Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 293, 14328–14341 (2018)
J.-S. Lee, J. Kang, S.-H. Lee, D. Hong, J. Son, K. Hong, J. Song, S.-Y. Kim, Dual targeting of glutaminase 1 and thymidylate synthase elicits death synergistically in NSCLC. Cell Death Dis. 7, e2511 (2016)
Y. Li, D.Y. Zhang, Q. Ren, F. Ye, X. Zhao, G. Daniels, X. Wu, B. Dynlacht, P. Lee, Regulation of a novel androgen receptor target gene, the cyclin B1 gene, through androgen-dependent E2F family member switching. Mol. Cell. Biol. 32, 2454–2466 (2012)
Y. Liu, T. Zhao, Z. Li, L. Wang, S. Yuan, L. Sun, The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 837, 81–87 (2018)
A. Luengo, D.Y. Gui, M.G. Vander Heiden, Targeting metabolism for cancer therapy. Cell. Chem. Biol. 24, 1161–1180 (2017)
C.E. Massie, A. Lynch, A. Ramos-Montoya, J. Boren, R. Stark, L. Fazli, A. Warren, H. Scott, B. Madhu, N. Sharma, H. Bon, V. Zecchini, D.M. Smith, G.M. Denicola, N. Mathews, M. Osborne, J. Hadfield, S. Macarthur, B. Adryan, S.K. Lyons, K.M. Brindle, J. Griffiths, M.E. Gleave, P.S. Rennie, D.E. Neal, I.G. Mills, The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 30, 2719–2733 (2011)
C.M. Metallo, P.A. Gameiro, E.L. Bell, K.R. Mattaini, J. Yang, K. Hiller, C.M. Jewell, Z.R. Johnson, D.J. Irvine, L. Guarente, J.K. Kelleher, M.G. Vander Heiden, O. Iliopoulos, G. Stephanopoulos, Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2011)
J. Munkley, Glycosylation is a global target for androgen control in prostate cancer cells. Endocr. Relat. Cancer 24, R49–r64 (2017)
J. Munkley, N.P. Lafferty, G. Kalna, C.N. Robson, H.Y. Leung, P. Rajan, D.J. Elliott, Androgen-regulation of the protein tyrosine phosphatase PTPRR activates ERK1/2 signalling in prostate cancer cells. BMC Cancer 15, 9 (2015)
J. Munkley, K.E. Livermore, U.L. McClurg, G. Kalna, B. Knight, P. McCullagh, J. McGrath, M. Crundwell, H.Y. Leung, C.N. Robson, L.W. Harries, P. Rajan, D.J. Elliott, The PI3K regulatory subunit gene PIK3R1 is under direct control of androgens and repressed in prostate cancer cells. Oncoscience 2, 755–764 (2015)
G.A. Nagana Gowda, G.A. Barding Jr., J. Dai, H. Gu, D.H. Margineantu, D.M. Hockenbery, D. Raftery, A metabolomics study of BPTES altered metabolism in human breast cancer cell lines. Front. Mol. Biosci. 5, 49 (2018)
M. Olsson, B. Zhivotovsky, Caspases and cancer. Cell. Death Differ. 18, 1441–1449 (2011)
J.A. Olzmann, P. Carvalho, Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell. Biol. 20, 137–155 (2019)
M. Ono, S. Oka, H. Okudaira, T. Nakanishi, A. Mizokami, M. Kobayashi, D.M. Schuster, M.M. Goodman, Y. Shirakami, K. Kawai, [(14)C]Fluciclovine (alias anti-[(14)C]FACBC) uptake and ASCT2 expression in castration-resistant prostate cancer cells. Nucl. Med. Biol. 42, 887–892 (2015)
T. Pan, L. Gao, G. Wu, G. Shen, S. Xie, H. Wen, J. Yang, Y. Zhou, Z. Tu, W. Qian, Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 456, 452–458 (2015)
C.F. Polotti, C.J. Kim, N. Chuchvara, A.B. Polotti, E.A. Singer, S. Elsamra, Androgen deprivation therapy for the treatment of prostate cancer: A focus on pharmacokinetics. Expert Opin. Drug. Metab. Toxicol. 13, 1265–1273 (2017)
X. Qu, J. Sun, Y. Zhang, J. Li, J. Hu, K. Li, L. Gao, L. Shen, c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem. Biophys. Res. Commun. 504, 415–421 (2018)
E.S. Reckzeh, G. Karageorgis, M. Schwalfenberg, J. Ceballos, J. Nowacki, M.C.M. Stroet, A. Binici, L. Knauer, S. Brand, A. Choidas, C. Strohmann, S. Ziegler, H. Waldmann, Inhibition of glucose transporters and Glutaminase synergistically impairs tumor cell growth. Cell Chem. Biol. 26, 1214–1228 (2019)
L.M.D. Reis, D. Adamoski, R. Ornitz Oliveira Souza, C.F. Rodrigues Ascencao, K.R. Sousa de Oliveira, F. Correa-da-Silva, F. Malta de Sa Patroni, M. Meira Dias, S.R. Consonni, P.M. Mendes de Moraes-Vieira, A.M. Silber, S.M.G. Dias, Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 294, 9342–9357 (2019)
M.M. Robinson, S.J. McBryant, T. Tsukamoto, C. Rojas, D.V. Ferraris, S.K. Hamilton, J.C. Hansen, N.P. Curthoys, Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 406, 407–414 (2007)
J.D. Schrag, J.J. Bergeron, Y. Li, S. Borisova, M. Hahn, D.Y. Thomas, M. Cygler, The structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol. Cell 8, 633–644 (2001)
M. Sciacovelli, C. Frezza, Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 284, 3132–3144 (2017)
L.G. Sheflin, W. Zhang, S.W. Spaulding, Androgen regulates the level and subcellular distribution of the AU-rich ribonucleic acid-binding protein HuR both in vitro and in vivo. Endocrinology 142, 2361–2368 (2001)
E.H. Shroff, L.S. Eberlin, V.M. Dang, A.M. Gouw, M. Gabay, S.J. Adam, D.I. Bellovin, P.T. Tran, W.M. Philbrick, A. Garcia-Ocana, S.C. Casey, Y. Li, C.V. Dang, R.N. Zare, D.W. Felsher, MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. U. S. A. 112, 6539–6544 (2015)
Z.A. Siddiqui, D.J. Krauss, Adjuvant androgen deprivation therapy for prostate cancer treated with radiation therapy. Transl. Androl. Urol. 7, 378–389 (2018)
D. Singh, T. Vishnoi, A. Kumar, Effect of alpha-ketoglutarate on growth and metabolism of cells cultured on three-dimensional cryogel matrix. Int. J. Biol. Sci. 9, 521–530 (2013)
Z. Song, B. Wei, C. Lu, P. Li, L. Chen, Glutaminase sustains cell survival via the regulation of glycolysis and glutaminolysis in colorectal cancer. Oncol. Lett. 14, 3117–3123 (2017)
K.R. Stone, D.D. Mickey, H. Wunderli, G.H. Mickey, D.F. Paulson, Isolation of a human prostate carcinoma cell line (DU 145). Int. J. Cancer 21, 274–281 (1978)
J. Teixeira, R. Amorim, K. Santos, P. Soares, S. Datta, G.A. Cortopassi, T.L. Serafim, V.A. Sardao, J. Garrido, F. Borges, P.J. Oliveira, Disruption of mitochondrial function as mechanism for anti-cancer activity of a novel mitochondriotropic menadione derivative. Toxicology 393, 123–139 (2018)
M.Y. Teo, D.E. Rathkopf, P. Kantoff, Treatment of advanced prostate cancer. Annu. Rev. Med. 70, 479–499 (2019)
R.M. Thompson, D. Dytfeld, L. Reyes, R.M. Robinson, B. Smith, Y. Manevich, A. Jakubowiak, M. Komarnicki, A. Przybylowicz-Chalecka, T. Szczepaniak, A.K. Mitra, B.G. Van Ness, M. Luczak, N.G. Dolloff, Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget 8, 35863–35876 (2017)
K. Vanhove, E. Derveaux, G.-J. Graulus, L. Mesotten, M. Thomeer, J.-P. Noben, W. Guedens, P. Adriaensens, Glutamine addiction and therapeutic strategies in lung cancer. Int. J. Mol. Sci. 20, 252 (2019)
C.V. Vaz, M.G. Alves, R. Marques, P.I. Moreira, P.F. Oliveira, C.J. Maia, S. Socorro, Androgen-responsive and nonresponsive prostate cancer cells present a distinct glycolytic metabolism profile. Int. J. Biochem. Cell Biol. 44, 2077–2084 (2012)
C.V. Vaz, R. Marques, M.G. Alves, P.F. Oliveira, J.E. Cavaco, C.J. Maia, S. Socorro, Androgens enhance the glycolytic metabolism and lactate export in prostate cancer cells by modulating the expression of GLUT1, GLUT3, PFK, LDH and MCT4 genes. J. Cancer Res. Clin. Oncol. 142, 5–16 (2016)
Q. Wang, R.A. Hardie, A.J. Hoy, M. van Geldermalsen, D. Gao, L. Fazli, M.C. Sadowski, S. Balaban, M. Schreuder, R. Nagarajah, J.J. Wong, C. Metierre, N. Pinello, N.J. Otte, M.L. Lehman, M. Gleave, C.C. Nelson, C.G. Bailey, W. Ritchie, J.E. Rasko, J. Holst, Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 236, 278–289 (2015)
Y. Wang, C. Bai, Y. Ruan, M. Liu, Q. Chu, L. Qiu, C. Yang, B. Li, Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 10, 201 (2019)
T. Wang, B. Cai, M. Ding, Z. Su, Y. Liu, L. Shen, c-Myc overexpression promotes oral cancer cell proliferation and migration by enhancing glutaminase and glutamine synthetase activity. Am. J. Med. Sci. 358, 235–242 (2019)
M.A. White, C. Lin, K. Rajapakshe, J. Dong, Y. Shi, E. Tsouko, R. Mukhopadhyay, D. Jasso, W. Dawood, C. Coarfa, D.E. Frigo, Glutamine transporters are targets of multiple oncogenic signaling pathways in prostate cancer. Mol. Cancer Res. 15, 1017–1028 (2017)
D.B. Williams, Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 119, 615–623 (2006)
Y. Xiang, Z.E. Stine, J. Xia, Y. Lu, R.S. O’Connor, B.J. Altman, A.L. Hsieh, A.M. Gouw, A.G. Thomas, P. Gao, L. Sun, L. Song, B. Yan, B.S. Slusher, J. Zhuo, L.L. Ooi, C.G. Lee, A. Mancuso, A.S. McCallion, A. Le, M.C. Milone, S. Rayport, D.W. Felsher, C.V. Dang, Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Invest. 125, 2293–2306 (2015)
D. Xu, M.E. Hemler, Metabolic activation-related CD147-CD98 complex. Mol. Cell Proteomics. 4, 1061–1071 (2005)
Y. Xu, S.Y. Chen, K.N. Ross, S.P. Balk, Androgens induce prostate cancer cell proliferation through mammalian target of rapamycin activation and post-transcriptional increases in cyclin D proteins. Cancer Res. 66, 7783–7792 (2006)
L. Yuan, X. Sheng, A.K. Willson, D.R. Roque, J.E. Stine, H. Guo, H.M. Jones, C. Zhou, V.L. Bae-Jump, Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. Relat. Cancer 22, 577–591 (2015)
L. Yuan, X. Sheng, L.H. Clark, L. Zhang, H. Guo, H.M. Jones, A.K. Willson, P.A. Gehrig, C. Zhou, V.L. Bae-Jump, Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am. J. Transl. Res. 8, 4265–4277 (2016)
N.M. Zacharias, C. McCullough, S. Shanmugavelandy, J. Lee, Y. Lee, P. Dutta, J. McHenry, L. Nguyen, W. Norton, L.W. Jones, P.K. Bhattacharya, Metabolic differences in glutamine utilization lead to metabolic vulnerabilities in prostate cancer. Sci. Rep. 7, 16159 (2017)
G. Zadra, C.F. Ribeiro, P. Chetta, Y. Ho, S. Cacciatore, X. Gao, S. Syamala, C. Bango, C. Photopoulos, Y. Huang, S. Tyekucheva, D.C. Bastos, J. Tchaicha, B. Lawney, T. Uo, L. D’Anello, A. Csibi, R. Kalekar, B. Larimer, L. Ellis, L.M. Butler, C. Morrissey, K. McGovern, V.J. Palombella, J.L. Kutok, U. Mahmood, S. Bosari, J. Adams, S. Peluso, S.M. Dehm, S.R. Plymate, M. Loda, Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 116, 631–640 (2019)
Y. Zhang, N. Fujita, T. Tsuruo, Caspase-mediated cleavage of p21Waf1/Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 18, 1131–1138 (1999)
J. Zhang, S. Mao, Y. Guo, Y. Wu, X. Yao, Y. Huang, Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 39, BSR20181826 (2019)
Acknowledgments
This work was supported by FEDER funds through the POCI - COMPETE 2020 - Operational Programme Competitiveness and Internationalization in Axis I -Strengthening research, technological development and innovation (Projects No. 007491 and 029114) and National Funds by the FCT-Foundation for Science and Technology (Project UID/Multi/00709/2013). Cardoso H. J. and Figueira M.I. are recipients of FCT fellowships SFRH/BD/111351/2015 and SFRH/BD/104671/2014, respectively. P.A.M. is funded by an FCT Investigator contract from the Foundation for Science and Technology (FCT), Portugal (Ref: IF/00614/2014) and an FCT exploratory grant, Ref: IF/00614/2014/CP12340006. CBMR is financed by an FCT Research Center grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Cardoso, H.J., Figueira, M.I., Vaz, C.V. et al. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5α-dihydrotestosterone regulation. Cell Oncol. 44, 385–403 (2021). https://doi.org/10.1007/s13402-020-00575-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-020-00575-9