
Abstract
We investigated the in vitro and in vivo osteogenic activity of licochalcone A. At low concentrations, licochalcone A stimulated the differentiation of mouse pre-osteoblastic MC3T3-E1 subclone 4 (MC4) cells and enhanced the bone morphogenetic protein (BMP)-2-induced stimulation of mouse bi-potential mesenchymal precursor C2C12 cells to commit to the osteoblast differentiation pathway. This osteogenic activity of licochalcone A was accompanied by the activation of extracellular-signal regulated kinase (ERK). The involvement of ERK was confirmed in a pharmacologic inhibition study. Additionally, noggin (a BMP antagonist) inhibited the osteogenic activity of licochalcone A in C2C12 cells. Licochalcone A also enhanced the BMP-2-stimulated expression of various BMP mRNAs. This suggested that the osteogenic action of licochalcone A in C2C12 cells could be dependent on BMP signaling and/or expression. We then tested the in vivo osteogenic activity of licochalcone A in two independent animal models. Licochalcone A accelerated the rate of skeletal development in zebrafish and enhanced woven bone formation over the periosteum of mouse calvarial bones. In summary, licochalcone A induced osteoblast differentiation with ERK activation in both MC4 and C2C12 cells and it exhibited in vivo osteogenic activity in zebrafish skeletal development and mouse calvarial bone formation. The dual action of licochalcone A in stimulating bone formation and inhibiting bone resorption, as described in a previous study, might be beneficial in treating bone-related disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone homeostasis is maintained by the bone remodeling process, which controls the balance between osteoclastic bone resorption and osteoblastic bone formation (Boyle et al. 2003; Harada and Rodan 2003). However, increased osteoclastic activity, decreased osteoblastic activity, or both can induce an imbalance in bone remodeling that consequently leads to a reduction in bone mass. Reduced bone mass increases the risk of fractures and the risk of developing metabolic bone diseases, like osteoporosis.
Many studies have sought to restore balance in bone remodeling by inhibiting osteoclastic bone resorption or enhancing osteoblastic bone formation. Others have attempted to treat bone diseases, including osteoporosis, by accelerating bone formation with anabolic agents alone or combined with anti-resorptive agents (Rosen and Bilezikian 2001; Garces and Garcia 2006). Also, several studies have identified active ingredients derived from natural substances that may be used to treat osteoporosis without adverse effects (Whelan et al. 2006; Putnam et al. 2007; Lee et al. 2010; Kim and Kim 2010).
Osteoblasts arise from mesenchymal stem cells and contribute to bone mineralization with activities like calcium deposition, which preserve bone strength and integrity (Katagiri et al. 1994). The cross-talk among several signaling pathways converges to regulate each step of osteoblast differentiation; in addition, mitogen-activated protein (MAP) kinases are essential for activating osteoblast differentiation via the induction of genes required for differentiation (Hipskind and Bilbe 1998). The earliest markers of osteoblasts, alkaline phosphatase (ALP) and collagen type I (Col1A1), and markers of mature osteoblasts, like osteopontin (OPN), have been shown to be controlled by the degree of cross-talk among MAP kinase pathways (Candeliere et al. 2001; Chae et al. 2002; Suzuki et al. 2002; Wu et al. 2006).
Licochalcone A (Fig. 1a) is derived from licorice, one of the most frequently used herbs in traditional medicine (Shibata 2000). It has been shown to have anti-inflammatory (Kolbe et al. 2006), anti-parasitic (Mi-Ichi et al. 2005), anti-cancer (Rafi et al. 2000; Fu et al. 2004), and anti-browning and depigmenting activities (Fu et al. 2005). Recently, we found that it exhibited anti-resorptive activity; at low concentrations (up to 5 μM), licochalcone A inhibited osteoclastogenesis and bone resorptive activity in mature osteoclasts without cytotoxicity (Kim et al. 2008a). This activity could alter bone homeostasis by changing the balance between osteoclastic bone resorption and osteoblastic bone formation; however, some phytochemicals have dual activities (Kim et al. 2008b, c). In this study, we investigated whether licochalcone A had an effect on osteoblast differentiation. We studied two cell lines, mouse pre-osteoblastic MC3T3-E1 subclone 4 (MC4) cells and bi-potential mesenchymal precursor C2C12 cells. We also examined the effect of licochalcone A on bone formation in two in vivo models: zebrafish skeletal development and mouse calvarial bone formation.

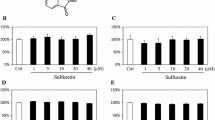
Structure of licochalcone A and its osteogenic activity in MC4 cells. a Chemical structure of licochalcone A. b Effect of licochalcone A on the viability of MC4 cells. The effect of licochalcone A on osteoblast differentiation was evaluated in MC4 cells by measuring (c) ALP activity and (d) its staining on day 9 of differentiation (described in “Materials and methods”). The effect of licochalcone A on mineralization was evaluated by measuring calcium deposits (e) and visualized by alizarin red S and von Kossa staining on days 15 and 18 of differentiation (f), respectively. *P < 0.001
Materials and methods
Materials
Licochalcone A and extracellular-signal regulated kinase (ERK) inhibitors, U0126 and PD98059, were purchased from Merck Biosciences Calbiochem (La Jolla, CA, USA) and Sigma (St Louis, MO, USA), respectively. Recombinant human bone morphogenetic protein (rhBMP)-2 and noggin were purchased from PeproTech (Seoul, Korea). All cell culture materials and antibodies were purchased from HyClone (Logan, UT, USA) and Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA), respectively.
Cell culture and differentiation
Mouse pre-osteoblastic MC3T3-E1 subclone 4 (MC4) cells and mouse bi-potential mesenchymal precursor C2C12 cells were purchased from American Type Culture Collection (Manassas, VA, USA). MC4 cells were cultured in growth medium (GM) with α-minimal essential medium (α-MEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 μg/ml streptomycin. Cells (1.5 × 104 cells/well) were plated in a 24-well plate and cultured in GM with humidified 5% CO2 at 37°C; the medium was changed every 3 days. After reaching confluence, the cells were cultured in differentiation medium (DM), which comprised GM with 50 μg/ml of ascorbic acid (Fluka, Germany) and 10 mM of β-glycerophosphate (Sigma); the medium was changed every 3 days. C2C12 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) that contained 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were seeded and, after 1 day, cells were differentiated by replacing the medium with DMEM with 5% FBS and rhBMP-2 (100 ng/ml). The medium was changed every 3 days.
Cell viability assay
MC4 cells were seeded in a 96-well plate in GM at 4 × 103 cells/well. After 24 h, cells were incubated in GM or DM with licochalcone A for 3 days. C2C12 cells were seeded in a 96-well plate at 4 × 103 cells/well. After 24 h, cells were incubated with licochalcone A for 1 or 3 days. Cell growth was then evaluated in triplicate with a Cell Counting Kit-8 (Dojindo Molecular Technologies, ML) according to the manufacturer’s protocol; absorbance was measured at 450 nm with the Wallac EnVision microplate reader (PerkinElmer, Finland); cell viability was presented as % of control (untreated).
Alkaline phosphatase staining and activity assay
Cells were washed twice with phosphate-buffered saline (PBS), fixed with 10% formalin in PBS for 30 s, rinsed with deionized water, and stained with an Alkaline Phosphatase (ALP) Kit (Sigma) under protection from direct light. Images of stained cells were captured under a microscope equipped with a DP70 digital camera (Olympus Optical, Tokyo, Japan). To measure ALP activity, cells were washed twice with PBS and sonicated in lysis buffer (10 mM of Tris–HCl, pH 7.5, 0.5 mM of MgCl2, and 0.1% Triton X-100). After centrifugation at 10,000×g for 20 min at 4°C, ALP activity in the supernatant was measured in triplicate with the LabAssay ALP Kit (Wako Pure Chemicals Industries, Osaka, Japan) according to the manufacturer’s protocol. ALP activity was presented in units per mg of cell lysate.
Alizarin red S staining
Cells were washed twice with PBS, stained with 40 mM alizarin red S solution (pH 4.2) for 10 min at room temperature, and washed twice with deionized water. Images of stained cells were captured under a microscope equipped with a DP70 digital camera.
Measurement of deposited calcium
To measure the amount of deposited calcium, cells were washed twice with PBS, fixed in 3.7% formaldehyde in PBS for 15 min, and decalcified with 300 μl of 1 N HCl for 24 h. Calcium concentration was measured with the Calcium C Kit (Wako Pure Chemicals Industries) according to the manufacturer’s protocol.
von Kossa staining
Cells were washed with PBS and fixed with 2.5% glutaraldehyde in PBS for 30 min. After washing three times with deionized water, cells were incubated with 5% silver nitrate at room temperature under UV light until the calcium turned black. After washing with deionized water three times, images of stained cells were captured with a microscope equipped with a DP70 digital camera.
Western blot analysis
Cells were homogenized in buffer that consisted of 10 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.05% (v/v) Tween 20, 1 mM phenylmethylsulfonyl fluoride (PMSF), and one protease inhibitor cocktail tablet (Roche, Mannheim, Germany) at 4°C. The homogenate was centrifuged at 10,000×g for 15 min. Concentrations of protein in the supernatant were determined with the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Samples (20 μg) were mixed with sample buffer (100 mM Tris–HCl, 2% sodium dodecyl sulfate, 1% 2-mercaptoethanol, 2% glycerol, 0.01% bromophenol blue, pH 7.6), incubated at 95°C for 15 min, and loaded onto 10% polyacrylamide gels. Electrophoresis was performed with the Mini Protean 3 Cell (Bio-Rad, Carlsbad, CA, USA). The resolved proteins were transferred to a nitrocellulose membrane (Schleicher & Schuell BioScience, Dassel, Germany). To ascertain the amount of protein loaded and the transfer efficiency, the membranes were stained with Ponceau S staining solution. For immunoanalysis, the membranes were washed and incubated in blocking buffer (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% Tween 20, and 3% nonfat dry milk) and then incubated with diluted primary antibodies (1:1,000) for 2 h at room temperature. Following the primary antibody reactions, the membranes were washed with blocking buffer three times (15 min each) and then probed with diluted secondary antibodies (1:2,000) for 1 h. The membranes were washed three times (15 min each), then developed with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce), and evaluated with the LAS-3000 luminescent image analyzer (Fuji Photo Film Co., Ltd., Kanagawa, Japan). The signal from a blot was quantified with ImageJ (http://rsb.info.nih.gov/ij/index.html) and presented as the fold change relative to the total intensity of the control.
Evaluation of mRNA expression
Primers (Table 1) were designed with an on-line primer design program (Rozen and Skaletsky 2000). Total RNA was isolated with TRIzol reagent (Life Technologies, Rockville, MD, USA) according to the manufacturer’s protocol. The concentration and purity of total RNA were calculated by measuring absorbance at 260 and 280 nm. First strand cDNA was synthesized with 2 μg of total RNA, 1 μM of oligo-dT18 primer, and Omniscript Reverse Transcriptase (Qiagen, Valencia, CA, USA). SYBR green-based quantitative PCR was performed with the Stratagene Mx3000P Real-Time PCR system and Brilliant SYBR Green Master Mix (Stratagene, La Jolla, CA, USA). Each reaction contained 3 μl of first-strand cDNA diluted 1:50 and 20 pmol of primers, according to the manufacturer’s protocols. The PCR thermocycling protocol consisted of three segments. The first segment (95°C for 10 min) activated the polymerase; the second segment included 40 cycles at 94°C for 40 s (denaturation), 60°C for 40 s (annealing), and 72°C for 1 min (extension); the third segment was performed to generate PCR product temperature dissociation curves (‘melting curves’) at 95°C for 1 min, 55°C for 30 s, and 95°C for 30 s. All reactions were run in triplicate, and data were analyzed with the 2−ΔΔCT method (Livak and Schmittgen 2001). A cDNA that encoded glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the control. Significance was determined with GAPDH-normalized 2−ΔΔCT values.
Alizarin red S-based zebrafish skeleton development assay
Zebrafish embryos were placed into a 24-well plate (7–10 embryos per well) 5 days after fertilization and maintained in 1 ml of buffered medium (sea salt, 0.06 mg/1L) that contained licochalcone A. The medium was changed every day. On day 8 post fertilization, the embryos were fixed in 4% paraformaldehyde and washed three times with PBS that contained 0.1% Tween 20 (PBST) at 10-min intervals. Next, the embryos were treated with a bleaching solution (PBST with 1% KOH and 3% H2O2) at room temperature until pigmented cells were removed (after about 30 min to 1 h). When the pigmented cell removal was complete, the embryos were washed 3 times with PBST at 10-min intervals and treated with 1 ml of alizarin red S staining buffer (pH 4.2) to stain the formed bone. After destaining in 1% KOH, the embryos were treated successively with KOH solutions that contained 20% glycerol, 50% glycerol, and 80% glycerol.
In vivo murine calvarial bone formation assay
The in vivo bone-forming activity of licochalcone A was evaluated with lyophilized collagen sponges, as described previously (Ha et al. 2006), with modifications. Briefly, collagen sponges were loaded with 5 μl of vehicle or 5 mM licochalcone A. The sponges were then implanted over the calvarial bones of ICR mice (n = 4 per group; Central Lab Animal, Seoul, Korea). Three weeks after drug implantation, the calvarias were harvested, fixed in 4% paraformaldehyde, decalcified in 12% EDTA, embedded in paraffin, and sectioned. Sections were deparaffinized through graded xylene washes, dehydrated in a graded series of ethanol washes, and stained with hematoxylin and eosin (H&E). The thickness of woven bones was quantified with the Image Pro-plus program, version 4.0 (Media Cybernetics, Inc., Bethesda, MD, USA).
Statistical analysis
Significance was determined using the Student’s t test and differences were considered significant when P < 0.05.
Results
Osteogenic activity of licochalcone A in MC4 cells
We evaluated the effect of licochalcone A (Fig. 1a) on osteoblast differentiation by measuring ALP activity in MC4 cells on day 9 of differentiation. Up to a concentration of 10 μM, licochalcone A did not exhibit any cytotoxicity under growth and differentiation culture conditions (Fig. 1b). Indeed, it significantly induced ALP activity at low concentrations (2–5 μM; Fig. 1c). This licochalcone A-stimulated ALP induction was visualized with ALP staining (Fig. 1d). We then investigated licochalcone A-induced mineralization by measuring the amounts of calcium deposited on days 15 and 18 of differentiation (Fig. 1e). Consistent with its stimulatory effect on ALP activity, licochalcone A significantly induced mineralization at low concentrations (2–5 μM); the level of deposited calcium in cells treated with licochalcone A was over 4-fold higher than that in the control. On differentiation day 18, calcium deposits were visualized by alizarin red S and von Kossa staining (Fig. 1f). Because we observed that licochalcone A had osteogenic activity at low concentrations, 5 μM licochalcone A was used in subsequent experiments.
We confirmed the osteogenic activity of licochalcone A in MC4 cells by measuring the mRNA expression levels of molecular markers for the early and late stages of osteoblastic differentiation; licochalcone A at 5 μM significantly induced the mRNA levels of ALP and Col1A1 on differentiation day 9 and the level of OPN on day 18 (Table 2).
ERK activation and AP-1 expression in licochalcone A-induced osteogenesis in MC4 cells
To elucidate the mechanism of action of licochalcone A in osteoblast differentiation, we used western blotting to evaluate its effect on MAP kinase activation. In MC4 cells, licochalcone A induced the phosphorylation of ERK within 5 min; conversely, it induced the dephosphorylation of JNK (Fig. 2a). These results suggested that ERK activation and/or JNK inactivation was involved in the osteogenic action of licochalcone A. To explore further the association between licochalcone A and MAP kinase activity, we evaluated the effect of the ERK-specific inhibitor, U0126, on licochalcone A-induced activation of MAP kinases and ALP expression (Fig. 2b). MC4 cells were treated with different concentrations of U0126 for 30 min and then incubated with licochalcone A for 5 min. This showed that U0126 dose-dependently inhibited licochalcone A-induced ERK activation. Additionally, U0126 functionally inhibited licochalcone A-stimulated ALP induction in a dose-dependent manner. We did not observe any cytotoxicity in MC4 cells associated with U0126 at the concentrations used in this study (data not shown). These results indicated that licochalcone A may affect osteogenesis via ERK activation.

Effects of licochalcone A on activation of signaling pathways. a The effect of licochalcone A on the activation of MAP kinases was evaluated by western blot analysis. MC4 cells (2 × 105 cells) were seeded in a 6-well plate and cultured in GM. After reaching confluence, cells were incubated with DM in the absence or presence of licochalcone A for 5–30 min. Western blot analysis was performed as described in “Materials and methods”. b Effect of U0126 on licochalcone A-stimulated ERK activation and ALP induction. MC4 cells (2 × 105 cells) were seeded in a 6-well plate and cultured in GM for 2 days. After reaching confluence, cells were pre-incubated with U0126 for 30 min and then incubated in DM with licochalcone A for 5 min. For ALP staining, cells (1.5 × 104 cells/well) were seeded in a 24-well plate and cultured in GM. After reaching confluence, cells were pre-incubated with U0126 for 2 h and then cultured in DM with licochalcone A. On day 3 of differentiation, licochalcone A was added after U0126 pretreatment for 2 h, and then ALP staining was performed on day 6. The blot signals were quantified with ImageJ and expressed as the fold change relative to the total intensity of the control
The induction of osteoblast differentiation-related genes could also be related to the induction of transcription factors; therefore, we investigated the effect of licochalcone A on the mRNA expression levels of activator protein (AP)-1 family members (c-Jun, JunD, c-Fos, Fra-1, and Fra-2) and the nuclear factor of activated T-cells, cytoplasmic, calcineurin-dependent 1 (NFATc1) with real-time PCR analysis. As shown in Table 3, on differentiation day 18, licochalcone A treatment was associated with significantly increased c-Jun, c-Fos, and Fra-1 mRNA expression levels.
ERK activation and BMP expression in licochalcone A-induced osteogenesis in C2C12 cells
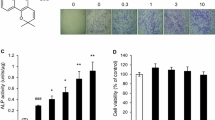
We next examined whether licochalcone A could induce bi-potential mesenchymal C2C12 cells to commit to the osteoblast differentiation pathway. Interestingly, licochalcone A exhibited osteogenic activity in C2C12 cells only in the presence of BMP-2. No cytotoxicity was observed at concentrations up to 5 μM of licochalcone A (Fig. 3a). Furthermore, in the presence of BMP-2 (200 ng/ml), licochalcone A stimulated ALP expression (Fig. 3b).

Osteogenic activity of licochalcone A and its mechanism of action in C2C12 cells. Effects of licochalcone A on cell viability (a), rhBMP-2-induced osteoblast differentiation (b), and activation of MAP kinases (c) were evaluated in C2C12 cells with a CCK-8 assay, ALP staining, and western blot analysis, respectively. For western blot analysis, cells (2 × 105 cells in a 6-well plate) were plated and cultured for 1 day. Cells were incubated in DMEM with 5% FBS, BMP-2, and licochalcone A for 5 min. The blot signals were quantified with ImageJ and expressed as the fold change relative to the total intensity of the control. d The effect of U0126 or PD98059 on licochalcone A-enhanced ERK activation and ALP induction in the presence of BMP-2 (100 ng/ml) was evaluated by western blot analysis and ALP staining/activity assay, respectively. The western blot analysis was performed and quantified as described above, but cells were pre-incubated with U0126 or PD98059 for 2 h. For ALP staining, cells (4 × 103 cells/well) were plated in a 96-well plate for 1 day. Cells were pre-incubated with U0126 or PD98059 for 2 h and then cultured in DMEM with 5% FBS, BMP-2 and licochalcone A for 2 days. e The effect of noggin on licochalcone A-enhanced ALP induction was evaluated by ALP staining, as described above. BMP-2 and licochalcone A treatment started on differentiation day 2, noggin treatment started on day 4, and ALP staining was performed on day 6
To elucidate the mechanism of action of licochalcone A in inducing C2C12 cells to commit to becoming osteoblasts, we evaluated MAP kinase activation in C2C12 cells (Fig. 3c). Consistent with the results in MC4 cells, licochalcone A dose-dependently induced the phosphorylation of ERK, but reduced that of JNK. We also used U0126 or PD98058 to experimentally confirm the involvement of ERK activation in the licochalcone A-induced enhancement of BMP-2 osteogenic activity. As shown in Fig. 3d, ERK activation and ALP induction by licochalcone A and BMP-2 could be inhibited by preincubation of the cells with U0126 or PD98059 for 2 h before treatment.
We reasoned that the ability of anabolic agents to stimulate mesenchymal stem cells to commit to becoming osteoblasts may depend on the cell’s ability to increase the level of endogenous BMPs. Therefore, we evaluated whether noggin, a BMP antagonist, inhibited licochalcone A-enhanced ALP induction in the presence of BMP-2. When C2C12 cells were incubated with BMP-2 and licochalcone A for 4 days and then incubated with noggin for an additional 2 days, licochalcone A-enhanced ALP induction was dose-dependently inhibited by noggin (Fig. 3e). Together with the BMP-2-dependent osteogenic activity of licochalcone A, these results suggested that BMP expression and signaling were involved in the osteogenic action of licochalcone A in C2C12 cells.
We further examined whether BMP expression was involved in the osteogenic action of licochalcone A in C2C12 cells by evaluating the mRNA expression levels of endogenous BMPs. As shown in Table 4, BMP-2 and BMP-4 mRNA expression levels were significantly increased by the combination of BMP-2 and licochalcone A and by BMP-2 alone. Additionally, BMP-7 and BMP-9 expression were also significantly induced by the combination of BMP-2 and licochalcone A. The mRNA induction of ALP (an early marker of osteoblast differentiation) and Runx2 (one of the major osteogenic transcription factors) were also confirmed in C2C12 cells by real-time PCR.
In vivo osteogenic activity of licochalcone A
Next, we examined the effect of licochalcone A on bone formation in two in vivo models, the zebrafish skeletal development model and the mouse calvarial bone formation model. In zebrafish embryos, at 8 days post fertilization, licochalcone A enhanced the development of Meckel’s cartilage (MC; a precursor of the temporomandibular joint), the palatoquadrate (pq), the opercle (op), and the vertebrae (vb), particularly at 5 μM (Fig. 4a). This corresponded with the results from the cell culture experiments. We also evaluated the in vivo bone-forming activity of licochalcone A with the mouse calvarial bone formation model. First, we implanted collagen sponges soaked with 5 μl of vehicle or 5 mM licochalcone A onto the calvarial bones of mice. After 3 weeks, the calvarial bones were examined for newly formed bone. H&E staining showed that licochalcone A significantly enhanced the woven bone formation over the periosteum of the calvarial bones (Fig. 4b); the thickness of newly formed woven bone in licochalcone A-treated mice was 3.5-fold higher than that in vehicle-treated mice.

Evaluation of in vivo osteogenic activity of licochalcone A in zebrafish and mouse calvaria. a Zebrafish at 5 days after fertilization were treated with licochalcone A for 3 days and then fixed and stained with alizarin red S. The appearance of Meckel’s cartilage (MC), palatoquadrate (pq), opercle (op), and vertebrae (vb) are indicated with arrows. b Collagen sponges containing licochalcone A (5 μl of 5 mM) were placed onto mouse calvarial bones. After 3 weeks, the mice were killed and calvarial bones were removed, fixed, decalcified, and embedded. Sections were stained with H&E and photographed at ×200 magnification. The arrows indicate the thickness of newly formed bone. The thickness of woven bones was quantified with the Image Pro-plus program. *P < 0.001
Discussion
In the present study, we found that licochalcone A exhibited osteogenic activity at low concentrations; it stimulated the differentiation of MC4 cells and enhanced the BMP-2-mediated induction of C2C12 cells to commit to becoming osteoblasts. Its osteogenic activity via ERK activation was confirmed, because it could be blocked with low concentrations of the ERK-specific inhibitor U0126 (≤0.3 μM) and PD98059 (≤0.1 μM). These results suggested that the ERK signaling pathway was involved in the osteogenic activity of licochalcone A. However, in contrast, the inhibition of ERK phosphorylation with high concentrations of PD98059 (≥25 μM) has been shown to promote the BMP-2-induced ALP activity in C2C12 cells (Gallea et al. 2001). This biphasic, concentration-dependent effect of ERK inhibitors like PD98059 should be evaluated in a future study. Furthermore, we confirmed the osteogenic activity of licochalcone A in two in vivo models. We found that licochalcone A enhanced skeletal development in zebrafish and significantly stimulated the formation of woven bone in mice.
In MC4 cells, 5 μM of licochalcone A significantly induced mRNA expression of ALP and Col1A1 on day 9 of differentiation and OPN expression on day 18. These genes are required for osteoblast differentiation and have been shown to be induced by the activation of MAP kinases. Previous studies suggested that p38 activation was critical for the regulation of ALP expression, one of the earliest markers of the osteoblast phenotype, during the differentiation of MC3T3-E1 cells (Suzuki et al. 2002). In primary human osteoblasts, ERK activation was directly related to ALP activity induced by a vitamin D3 analog (Chae et al. 2002). Furthermore, Col1A1 expression during matrix maturation was regulated by both ERK and JNK signaling pathways (Wu et al. 2006). Finally, mineralization results in the induction of several non-collagenous proteins that provide markers for mature osteoblasts, including OPN (Candeliere et al. 2001). Interestingly, the oscillatory flow-activated induction of OPN expression was reported to be mediated through the ERK pathway (Wu et al. 2006). Taking these findings into consideration, the complexity of osteoblastic gene expression could be controlled by the degree of cross-talk between MAP kinase pathways. In turn, the activation of each MAP kinase involved in the process of osteoblast differentiation is likely to be dependent upon the cell type, the particular stimuli, or the stage of differentiation. In this study, licochalcone A triggered the activation of ERK in MC4 cells under differentiation conditions. Furthermore, licochalcone A-induced ERK activation and ALP induction were dose-dependently inhibited by U0126, an ERK-specific inhibitor. This suggested that the stimulatory effect of licochalcone A on the differentiation of MC4 cells could depend on its potential to activate the ERK signaling pathway.
Additionally, the MAP kinase induction of the expression of genes required for osteoblast differentiation could be regulated by the coordination between MAP kinases and transcription factors like AP-1 and NFATc1. For example, uridine triphosphate-induced OPN expression is mediated by the coordination between the protein kinase C-activated NF-κB pathway and the ERK-activated AP-1 pathway (Renault et al. 2005). Furthermore, several studies have suggested that induction of AP-1 expression could control bone formation and remodeling by regulating the expression of several osteoblast-specific genes through AP-1 binding sites in their promoters (McCabe et al. 1996; Wagner 2002; Narayanan et al. 2004; Sakata et al. 2004). In this study, licochalcone A at 5 μM significantly induced the mRNA expression levels of several AP-1 family members, including c-Jun, c-Fos, and Fra-1, but not NFATc1. This suggested that AP-1 expression could be involved in the osteogenic activity of licochalcone A in MC4 cells. Our results are consistent with those of other studies that reported extracellular glucose-induced up-regulation of c-Jun expression and transcriptional induction of c-Fos expression by parathyroid hormone during osteoblast differentiation (McCauley et al. 1997; Zayzafoon et al. 2000). Furthermore, Fra-1 was shown to be an important regulator of bone mass; it affected bone matrix production and maintained osteoblast activity (Jochum et al. 2000; Eferl et al. 2004).
BMPs control the initial differentiation of mesenchymal stem cells (MSCs) into osteoblasts. Previous studies have shown that BMPs could potently induce osteogenesis (Phimphilai et al. 2006) and that MAP kinases were involved in the BMP-2-induced stimulation of bi-potential C2C12 cells to commit to the osteoblast pathway (Gallea et al. 2001). In our study, licochalcone A induced ALP expression and ERK activation in C2C12 cells in the presence of BMP-2. Moreover, these inductions were inhibited by U0126. This suggested that the effect of licochalcone A on the BMP-2-induced commitment to the osteoblast pathway may have depended on its potential to induce ERK activation. These results were consistent with those observed in MC4 cells. In addition, the stimulatory effect of licochalcone A on ALP expression was not observed in the absence of BMP-2 (data not shown); furthermore, treatment with noggin dose-dependently inhibited the licochalcone A-mediated enhancement of BMP-2-stimulated ALP induction. These results suggested that the osteogenic activity of licochalcone A might be dependent on BMP signaling and expression.
We then evaluated the osteogenic action of licochalcone A by examining its potential to induce the expression of endogenous BMPs. Real-time PCR revealed that licochalcone A significantly enhanced the BMP-2-induced mRNA expression of BMP-2, BMP-4, BMP-7, and BMP-9. The osteogenic activity of BMP-2 was previously reported in human clinical trials (Boden et al. 2000; Valentin-Opran et al. 2002); the osteogenic activities of BMP-4, BMP-7, and BMP-9 were reported in C2C12 cells (Yeh et al. 2002; Li et al. 2003; Bessa et al. 2009). The ability of BMPs to enhance the expression of other BMP genes could play an important role in the process of bone formation. Gene transfer of BMP-2 and BMP-7 induced rapid bone formation and increased endogenous BMP-4 expression (Kawai et al. 2006). BMP-2 treatment enhanced the expression of other BMP genes during bone cell differentiation (Chen et al. 1997). Additionally, in our study, licochalcone A enhanced the BMP-2-stimulated induction of Runx2 mRNA expression. Runx2 is a key transcription factor in the BMP-2 signaling pathway, which controls osteoblast precursor cell differentiation by regulating the expression of osteogenesis-related genes (Lian et al. 2003; Gersbach et al. 2004; Phimphilai et al. 2006; Bessa et al. 2009).
Additionally, in both MC4 and C2Cl2 cells, JNK was inhibited during licochalcone A-induced osteoblast differentiation. TGF-β-responsive JNK cascades have been suggested to negatively regulate ALP activity and mineralization in MC3T3-E1 cells (Sowa et al. 2002). Furthermore, TNF-α inhibited the BMP-induced osteoblast differentiation in C2C12 cells by activating JNK signaling; a pharmacological study revealed that the JNK inhibitor, SP600125, partially restored the TNF-α-mediated suppression of BMP-induced Runx2 and osteocalcin mRNA expression (Mukai et al. 2007). Those results suggested that JNK inhibition might be involved in activating the osteogenic process in both MC4 and C2C12 cells.
The observed in vitro osteogenic activity of licochalcone A led us to evaluate its in vivo osteogenic activity with animal models. Zebrafish were previously demonstrated to be successful models for identifying agents with anabolic effects on the skeleton (Du et al. 2001; Fleming et al. 2005). When zebrafish embryos were treated with 5 μM licochalcone A, the rate of skeletal development accelerated; this observation corresponded with the results of our cell culture experiments. We also confirmed the in vivo bone-forming activity of licochalcone A in a mouse calvarial bone formation model. When collagen sponges soaked with licochalcone A were implanted onto the calvarial bones of mice, woven bone formation was significantly enhanced over the periosteum of the calvarial bones. In these experiments, we did not add any BMPs with licochalcone A; nevertheless, new bone was formed. Considering that subcutaneous injection of BMP-2 has been shown to stimulate periosteal surface bone formation in mice (Chen et al. 1997), licochalcone A may trigger bone formation by inducing the activation of endogenous BMPs in mice. Additionally, considering that licochalcone A could induce BMP mRNA expression, we cannot rule out the possibility that the in vivo osteogenic activity of licochalcone A in mice might have been due to its induction of BMP expression.
In conclusion, we found that licochalcone A exhibited osteogenic activity in vitro and in vivo. Licochalcone A significantly induced osteoblast differentiation through ERK activation in MC4 and C2C12 cells; more importantly, it showed in vivo activity in zebrafish skeletal development and mouse calvarial bone formation models. The dual action of licochalcone A in stimulating bone formation and inhibiting bone resorption (Kim et al. 2008a) might be beneficial for treating bone-related disorders.
References
Bessa PC, Cerqueira MT, Rada T, Gomes ME, Neves NM, Nobre A, Reis RL, Casal M (2009) Expression, purification and osteogenic bioactivity of recombinant human BMP-4, -9, -10, -11 and -14. Protein Expr Purif 63(2):89–94
Boden SD, Zdeblick TA, Sandhu HS, Heim SE (2000) The use of rhBMP-2 in interbody fusion cages. Definitive evidence of osteoinduction in humans: a preliminary report. Spine 25(3):376–381
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423(6937):337–342
Candeliere GA, Liu F, Aubin JE (2001) Individual osteoblasts in the developing calvaria express different gene repertoires. Bone 28(4):351–361
Chae HJ, Jeong BJ, Ha MS, Lee JK, Byun JO, Jung WY, Yun YG, Lee DG, Oh SH, Chae SW, Kwak YG, Kim HH, Lee ZH, Kim HR (2002) ERK MAP kinase is required in 1, 25(OH)2D3-induced differentiation in human osteoblasts. Immunopharmacol Immunotoxicol 24(1):31–41
Chen D, Harris MA, Rossini G, Dunstan CR, Dallas SL, Feng JQ, Mundy GR, Harris SE (1997) Bone morphogenetic protein 2 (BMP-2) enhances BMP-3, BMP-4, and bone cell differentiation marker gene expression during the induction of mineralized bone matrix formation in cultures of fetal rat calvarial osteoblasts. Calcif Tissue Int 60(3):283–290
Du SJ, Frenkel V, Kindschi G, Zohar Y (2001) Visualizing normal and defective bone development in zebrafish embryos using the fluorescent chromophore calcein. Dev Biol 238(2):239–246
Eferl R, Hoebertz A, Schilling AF, Rath M, Karreth F, Kenner L, Amling M, Wagner EF (2004) The Fos-related antigen Fra-1 is an activator of bone matrix formation. EMBO 23(14):2789–2799
Fleming A, Sato M, Goldsmith P (2005) High-throughput in vivo screening for bone anabolic compounds with zebrafish. J Biomol Screen 10(8):823–831
Fu Y, Hsieh TC, Guo J, Kunicki J, Lee MY, Darzynkiewicz Z, Wu JM (2004) Licochalcone-A, a novel flavonoid isolated from licorice root (Glycyrrhiza glabra), causes G2 and late-G1 arrests in androgen-independent PC-3 prostate cancer cells. Biochem Biophys Res Commun 322(1):263–270
Fu B, Li H, Wang X, Lee FS, Cui S (2005) Isolation and identification of flavonoids in licorice and a study of their inhibitory effects on tyrosinase. J Agric Food Chem 53(19):7408–7414
Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S, Kawai S, Faucheu C, Huet L, Baron R, Roman-Roman S (2001) Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone 28(5):491–498
Garces C, Garcia LE (2006) Combination of anabolic and antiresorptive agents for the treatment of osteoporosis. Maturitas 54(1):47–54
Gersbach CA, Byers BA, Pavlath GK, Garcia AJ (2004) Runx2/Cbfa1 stimulates transdifferentiation of primary skeletal myoblasts into a mineralizing osteoblastic phenotype. Exp Cell Res 300(2):406–417
Ha H, Lee JH, Kim HN, Kim HM, Kwak HB, Lee S, Kim HH, Lee ZH (2006) α-Lipoic acid inhibits inflammatory bone resorption by suppressing prostaglandin E2 synthesis. J Immunol 176(1):111–117
Harada S, Rodan GA (2003) Control of osteoblast function and regulation of bone mass. Nature 423(6937):349–355
Hipskind RA, Bilbe G (1998) MAP kinase signaling cascades and gene expression in osteoblasts. Front Biosci 3:d804–d816
Jochum W, David JP, Elliott C, Wutz A, Plenk H Jr, Matsuo K, Wagner EF (2000) Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat Med 6(9):980–984
Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, Rosen V, Wozney JM, Fujisawa-Sehara A, Suda T (1994) Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol 127(6 Pt 1):1755–1766
Kawai M, Bessho K, Maruyama H, Miyazaki J, Yamamoto T (2006) Simultaneous gene transfer of bone morphogenetic protein (BMP)-2 and BMP-7 by in vivo electroporation induces rapid bone formation and BMP-4 expression. BMC Musculoskelet Disord 7:62
Kim HJ, Kim SH (2010) Tanshinone IIA enhances BMP-2-stimulated commitment of C2C12 cells into osteoblasts via p38 activation. Amino Acids 39(5):1217–1226
Kim SN, Kim MH, Min YK, Kim SH (2008a) Licochalcone A inhibits the formation and bone resorptive activity of osteoclasts. Cell Biol Int 32(9):1064–1072
Kim MH, Ryu SY, Bae MA, Choi JS, Min YK, Kim SH (2008b) Baicalein inhibits osteoclast differentiation and induces mature osteoclast apoptosis. Food Chem Toxicol 46(11):3375–3382
Kim JM, Lee SU, Kim YS, Min YK, Kim SH (2008c) Baicalein stimulates osteoblast differentiation via coordinating activation of MAP kinases and transcription factors. J Cell Biochem 104(5):1906–1917
Kolbe L, Immeyer J, Batzer J, Wensorra U, Dieck KT, Mundt C, Wolber R, Stab F, Schonrock U, Ceilley RI, Wenck H (2006) Anti-inflammatory efficacy of licochalcone A: correlation of clinical potency and in vitro effects. Arch Dermatol Res 298(1):23–30
Lee SU, Kang NS, Min YK, Kim SH (2010) Camphoric acid stimulates osteoblast differentiation and induces glutamate receptor expression. Amino Acids 38(1):85–93
Li JZ, Li H, Sasaki T, Holman D, Beres B, Dumont RJ, Pittman DD, Hankins GR, Helm GA (2003) Osteogenic potential of five different recombinant human bone morphogenetic protein adenoviral vectors in the rat. Gene Ther 10(20):1735–1743
Lian JB, Stein JL, Stein GS, van Wijnen AJ, Montecino M, Javed A, Gutierrez S, Shen J, Zaidi SK, Drissi H (2003) Runx2/Cbfa1 functions: diverse regulation of gene transcription by chromatin remodeling and co-regulatory protein interactions. Connect Tissue Res 44(Suppl 1):141–148
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25(4):402–408
McCabe LR, Banerjee C, Kundu R, Harrison RJ, Dobner PR, Stein JL, Lian JB, Stein GS (1996) Developmental expression and activities of specific fos and jun proteins are functionally related to osteoblast maturation: role of Fra-2 and Jun D during differentiation. Endocrinology 137(10):4398–4408
McCauley LK, Koh AJ, Beecher CA, Rosol TJ (1997) Proto-oncogene c-fos is transcriptionally regulated by parathyroid hormone (PTH) and PTH-related protein in a cyclic adenosine monophosphate-dependent manner in osteoblastic cells. Endocrinology 138(12):5427–5433
Mi-Ichi F, Miyadera H, Kobayashi T, Takamiya S, Waki S, Iwata S, Shibata S, Kita K (2005) Parasite mitochondria as a target of chemotherapy: inhibitory effect of licochalcone A on the Plasmodium falciparum respiratory chain. Ann NY Acad Sci 1056:46–54
Mukai T, Otsuka F, Otani H, Yamashita M, Takasugi K, Inagaki K, Yamamura M, Makino H (2007) TNF-alpha inhibits BMP-induced osteoblast differentiation through activating SAPK/JNK signaling. Biochem Biophys Res Commun 356(4):1004–1010
Narayanan K, Srinivas R, Peterson MC, Ramachandran A, Hao J, Thimmapaya B, Scherer PE, George A (2004) Transcriptional regulation of dentin matrix protein 1 by JunB and p300 during osteoblast differentiation. J Biol Chem 279(43):44294–44302
Phimphilai M, Zhao Z, Boules H, Roca H, Franceschi RT (2006) BMP signaling is required for RUNX2-dependent induction of the osteoblast phenotype. J Bone Miner Res 21(4):637–646
Putnam SE, Scutt AM, Bicknell K, Priestley CM, Williamson EM (2007) Natural products as alternative treatments for metabolic bone disorders and for maintenance of bone health. Phytother Res 21(2):99–112
Rafi MM, Rosen RT, Vassil A, Ho CT, Zhang H, Ghai G, Lambert G, DiPaola RS (2000) Modulation of bcl-2 and cytotoxicity by licochalcone-A, a novel estrogenic flavonoid. Anticancer Res 20(4):2653–2658
Renault MA, Jalvy S, Potier M, Belloc I, Genot E, Dekker LV, Desgranges C, Gadeau AP (2005) UTP induces osteopontin expression through a coordinate action of NFkappaB, activator protein-1, and upstream stimulatory factor in arterial smooth muscle cells. J Biol Chem 280(4):2708–2713
Rosen CJ, Bilezikian JP (2001) Clinical review 123: anabolic therapy for osteoporosis. J Clin Endocrinol Metab 86(3):957–964
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Sakata R, Minami S, Sowa Y, Yoshida M, Tamaki T (2004) Trichostatin A activates the osteopontin gene promoter through AP1 site. Biochem Biophys Res Commun 315(4):959–963
Shibata S (2000) A drug over the millennia: pharmacognosy, chemistry, and pharmacology of licorice. Yakugaku Zasshi 120(10):849–862
Sowa H, Kaji H, Yamaguchi T, Sugimoto T, Chihara K (2002) Activations of ERK1/2 and JNK by transforming growth factor beta negatively regulate Smad3-induced alkaline phosphatase activity and mineralization in mouse osteoblastic cells. J Biol Chem 277(39):36024–36031
Suzuki A, Guicheux J, Palmer G, Miura Y, Oiso Y, Bonjour JP, Caverzasio J (2002) Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone 30(1):91–98
Valentin-Opran A, Wozney J, Csimma C, Lilly L, Riedel GE (2002) Clinical evaluation of recombinant human bone morphogenetic protein-2. Clin Orthop Relat Res 395:110–120
Wagner EF (2002) Functions of AP1 (Fos/Jun) in bone development. Ann Rheum Dis 61(Suppl 2):ii40–ii42
Whelan AM, Jurgens TM, Bowles SK (2006) Natural health products in the prevention and treatment of osteoporosis: systematic review of randomized controlled trials. Ann Pharmacother 40(5):836–849
Wu CC, Li YS, Haga JH, Wang N, Lian IY, Su FC, Usami S, Chien S (2006) Roles of MAP kinases in the regulation of bone matrix gene expressions in human osteoblasts by oscillatory fluid flow. J Cell Biochem 98(3):632–641
Yeh LC, Tsai AD, Lee JC (2002) Osteogenic protein-1 (OP-1, BMP-7) induces osteoblastic cell differentiation of the pluripotent mesenchymal cell line C2C12. J Cell Biochem 87(3):292–304
Zayzafoon M, Stell C, Irwin R, McCabe LR (2000) Extracellular glucose influences osteoblast differentiation and c-Jun expression. J Cell Biochem 79(2):301–310
Acknowledgments
CHK was supported by a grant from the National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (1020090). This study was supported by a grant from the National R&D program of the Korea Ministry of Education, Science, and Technology, Republic of Korea (Chemical Genomics Research, 20100002073).
Author information
Authors and Affiliations
Corresponding authors
Additional information
S. H. Kim and C. H. Kim are corresponding authors. S. N. Kim and S. J. Bae contributed equally to this study.
Rights and permissions
About this article
Cite this article
Kim, S.N., Bae, S.J., Kwak, H.B. et al. In vitro and in vivo osteogenic activity of licochalcone A. Amino Acids 42, 1455–1465 (2012). https://doi.org/10.1007/s00726-011-0901-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-011-0901-7