
Abstract
Bone morphogenetic protein 2 (BMP-2) has been used clinically to encourage bone regeneration; although, there can be major side effects with larger doses. Therefore, there is a need to identify new small molecules to potentiate the osteogenic action of BMP-2. In this study, we investigated the effect of mollugin on bone formation in murine bi-potential mesenchymal progenitor C2C12 cells by combination with BMP-2. We found mollugin could enhance the BMP-2-mediated osteoblast differentiation of C2C12 cells. This was accompanied by the induction of other osteogenic BMPs. We also found the enhancing potential of mollugin may involve activation of the p38–Smad1/5/8 signaling axis. Furthermore, mollugin promoted skeletal development in zebrafish. The combination of BMP-2 with small molecules, including mollugin, could minimize its clinical limitations, and these molecules might lead to the development of effective stem cell stimulants for bone regeneration and fracture healing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bone morphogenetic proteins (BMPs) are most potent growth factors that promote osteogenesis (Cheng et al. 2003). Commercially available BMP-2 has been used as a clinical therapy for spinal injuries, bone grafts, and other orthopedic surgeries to encourage bone regeneration in patients (McKay et al. 2007; Benglis et al. 2008; Irad and Einhorn 2009). Recombinant human BMP-2 (1.5 mg/ml) has enhanced bone repair in patients with open tibia fractures in a human trial study (Alt et al. 2009). Globally, the percentage of the elderly that require various bone-related healthcare products continues to grow. The “Transparency Market Research” report expects the global BMP market to rise up to $587 million by 2022. However, the clinical application of BMP-2 in high doses can cause life-threating side effects, including cervical spine swelling or even tumors (James et al. 2016). Thus, there is a need to identify small molecules to enhance the osteogenic action of BMPs. The combination of BMP-2 and these small molecules could lower the doses of BMP-2, and thus, the risk of its side effects and the potential cost of BMP-2-based clinical treatment in the future (Cao et al. 2014; Fan et al. 2016).
Natural products are promising resources for identifying bioactive molecules. Since natural products provide novel molecular scaffolds and chemistry, many therapeutics including anti-cancer drugs are natural product-based/inspired (Mondal et al. 2012). In fact, in the field of bone biology, many bioactive natural products have been identified (Weaver et al. 2012), and several phytochemicals enhance the osteogenic action of BMP-2. More interestingly, phytochemicals, such as licochalcone A and tanshinone IIA, also exhibited the anti-osteoclastogenic activity (Kwak et al. 2006; Kim et al. 2008, 2012; Kim and Kim 2010).
Mollugin (naphthohydroquinone; Fig. 1a) is a non-mutagenic constituent isolated from the root of Rubia tinctorum in Europe, Rubia akane in Taiwan, or Rubia cordifolia in India (Kawasaki et al. 1992). It has exhibited several biological activities including neuroprotective (Jeong et al. 2011), anti-viral (Ho et al. 1996), anti-inflammatory (Kim et al. 2009; Zhu et al. 2013), anti-adipogenic (Jun et al. 2011), and anti-cancer activities (Do et al. 2013; Lee et al. 2013). Interestingly, anti-osteoclastogenic activity of mollugin was recently reported (Baek et al. 2015), but its action in bone formation has not yet been evaluated. In this study, the effect of mollugin on bone formation was investigated in murine bi-potential mesenchymal progenitor C2C12 cells by combination with BMP-2. The BMP-2-induced commitment of C2C12 cells into osteoblasts is a well-defined in vitro bone formation model.

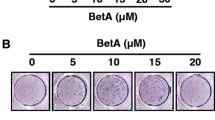
Mollugin enhances BMP-2-induced ALP. a Chemical structure of mollugin. The effect of mollugin on BMP-2-induced osteoblast differentiation was evaluated by visualizing ALP staining b and measuring ALP activity c in C2C12 cells. d The effect of mollugin on the viability of C2C12 cells was evaluated using the CCK-8 assay. DMSO (0.1%) was used as the vehicle control. ## p < 0.01 (vs the vehicle control group); *p < 0.05; **p < 0.01 (vs the BMP-2-treated group)
Materials and methods
Materials
Mollugin was synthesized as described previously (Lee et al. 2007). Recombinant human BMP-2 (rhBMP-2) and SB202190 (p38 inhibitor) were purchased from R&D Systems (Minneapolis, MN, USA) and Calbiochem (San Diego, CA, USA), respectively. All cell culture materials were purchased from HyClone (Logan, UT, USA). Antibodies against phospho(p)-p38, p38, p–c-Jun N-terminal kinase (p-JNK), JNK, p-extracellular-signal-regulated kinase (p-ERK), ERK, p-Sma and Mad related proteins 1/5/8 (p-Smad1/5/8), and Smad were purchased from Cell Signaling Technology Inc. (Danvers, MA, USA). The antibody against actin and the secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
Cell culture and differentiation
The C2C12 cells were purchased from the American Type Culture Collection (Manassas, VA, USA) and were maintained in growth medium (GM; Dulbecco’s Modified Eagle’s Medium [DMEM] containing 10% fetal bovine serum [FBS], 100 U/ml of penicillin, and 100 μg/ml streptomycin). Cells were seeded and, after 1 day, differentiated by replacing the medium with differentiation medium (DM; DMEM containing 5% FBS and 50 ng/ml rhBMP-2).
Alkaline phosphatase (ALP) staining and activity assay
Alkaline phosphatase (ALP), an early biomarker of osteoblastogenesis, was assayed as described previously (Kim and Kim 2010). Briefly, C2C12 cells (4 × 103 cells/well) were seeded in a 96-well plate, and after 24 h, the medium was replaced with DM in the absence or presence of mollugin. After 3 days, cells were washed twice with phosphate-buffered saline (PBS), fixed with 10% formalin in PBS for 30 s, rinsed with deionized water, and stained using the Alkaline Phosphatase Kit (Sigma, St. Louis, MO, USA). To measure ALP activity, cells were washed twice with PBS, and 100 μl of the 1-Step PNPP (p-nitrophenyl phosphate) substrate solution (Thermo Fisher Scientific, Waltham, MA, USA) was added to each well. Then, cells were incubated for 15 min at room temperature and the reaction was stopped by adding 50 μl of 2 N NaOH. Then, absorbance was measured at 405 nm using a Wallac EnVision microplate reader (PerkinElmer, Waltham, MA, USA).
Cell viability assay
Cells were seeded in a 96-well plate at 4 × 103 cells/well. After 24 h, cells were incubated with mollugin for 3 days, and then cell viability was evaluated in triplicate using a Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer’s protocol. Absorbance was measured at 450 nm using a Wallac EnVision microplate reader.
Evaluation of mRNA expression
The mRNA expression level was evaluated as described previously (Kim and Kim 2010). Briefly, primers used in this study were designed using an online primer design program (Rozen and Skaletsky 2000; Table 1). Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific) and the first strand cDNA was synthesized using 1 μg of total RNA, 1 μM of oligo-dT18 primer, 10 units of the RNase inhibitor, RNasin (Promega, Madison, WI, USA), and Omniscript Reverse Transcriptase (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. Then, a SYBR green-based quantitative polymerase chain reaction (PCR) was performed using the Stratagene Mx3000P Real-Time PCR system and Brilliant SYBR Green Master Mix (Stratagene, La Jolla, CA, USA) in a 20 μl reaction mixture containing 10 μl of TOPreal qPCR 2× PreMIX (Enzynomics, Daejeon, Korea), 2 pmol of forward primer, 2 pmol of reverse primer, and 2 μl of 1:10-diluted first strand cDNA. Amplification parameters consisted of an initial denaturation at 95 °C for 10 min and 40 cycles of three-step PCR (denaturation at 94 °C for 40 s, annealing at 53 °C for 40 s, and extension at 72 °C for 1 min). All reactions were run in triplicate and data were analyzed using the 2−∆∆CT method (Livak and Schmittgen 2001). Glyceraldehye 3-phosphate dehydrogenase (GAPDH) was used as the control gene. Significance was determined with GAPDH-normalized 2−∆∆CT values.
Western blot analysis
Cells were lysed with cold lysis buffer (0.2 M Tris–HCl, pH 7.5, 150 mM NaCl, 1% deoxycholic acid, 1% NP-40, and 1 mM EDTA) containing 1× protease and phosphatase inhibitors (Pierce Biotechnology, Rockford, IL, USA). Cell lysates were subsequently incubated at 4 °C for 20 min and then centrifuged at 14,000×g for 15 min. The protein concentration of the supernatants was determined using a protein assay kit (Bio-Rad, Hercules, CA, USA). Protein samples (20 μg) in sample buffer (100 nM Tris–HCl, 2% sodium dodecyl sulfate (SDS), 1% 2-mercaptoethanol, 2% glycerol, 0.01% bromophenol blue, pH 7.6) were denatured at 95 °C for 15 min and loaded on 10% polyacrylamide gels. Electrophoresis was performed with the Mini Protean 3 Cell (Bio-Rad) and the resolved proteins were transferred by electroblotting to polyvinylidene difluoride (PVDF) membrane (Millipore, Temecula, CA, USA). Membranes were incubated with TBST buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween 20) with 5% nonfat dry milk and incubated with diluted primary antibodies (1:1000) overnight at 4 °C. The membranes were washed with TBST at room temperature and incubated with diluted secondary antibodies (1:5000) for 1 h. After washing with TBST three times (15 min each), the membranes were developed with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology) and evaluated with the LAS-3000 luminescent image analyzer (Fuji Photo Film Co., Ltd., Tokyo, Japan).
Zebrafish skeleton development
As described previously (Moon et al. 2015), the skeleton development of zebrafish was visualized by alizarin red S according to the standard protocol approved by the Institutional Animal Care and Use Committees of Chungnam National University (CNU-00393). Briefly, zebrafish embryos were added to a 24-well plate (5/well) 5 days after fertilization and maintained in 1 ml of buffered medium (sea salt, 0.06 mg/l) with or without mollugin. The medium was changed every day. At 8 days after fertilization, embryos were fixed in 4% paraformaldehyde for 1 h, washed 3 times with PBS containing 0.1% Tween 20 (PBST) at 10 min intervals, and treated with a bleaching solution (PBST with 1% KOH and 3% H2O2) at room temperature until pigmented cells were removed. After the pigmented cell removal was complete, embryos were washed 3 times with PBST at 10 min intervals and treated with 1 ml of alizarin red S staining buffer (pH 4.2). After destaining with 1% KOH, embryos were treated successively with KOH solutions that contained 20% glycerol, 50% glycerol, and 80% glycerol. Images were acquired using a Leica microscope (Wetzlar, Germany).
Statistical analysis
All quantitative values are presented as the mean ± SD. Each experiment was performed three times, and results from one representative experiment are presented here. Statistical differences were analyzed using Student’s t test. A value of p < 0.05 was considered significant.
Results
Mollugin enhances BMP-2-induced ALP
Biological activity of mollugin in the process of bone formation was evaluated in C2C12 cells by ALP staining and its activity measurement. Since mollugin per se did not show any osteogenic action without BMP-2 (data not shown), its enhancing effect on the BMP-2-induced differentiation of C2C12 cells was evaluated. As shown in Fig. 1b and c, 50 ng/ml of BMP-2 significantly induced the activity of ALP, the representative marker for osteoblast differentiation, and its induction was enhanced by mollugin in a dose-dependent manner. Up to 10 μM, mollugin did not exhibit any cytotoxicity under the growth culture conditions (Fig. 1d). In addition, mollugin significantly enhanced the BMP-2-induced mRNA expression of ALP at 3–10 μM (Fig. 2).

Mollugin enhances BMP-2-induced mRNA expression of ALP and osteogenic BMPs in C2C12 cells. Cells were treated with the vehicle (0.1% DMSO), BMP-2 (50 ng/ml), or BMP-2 and mollugin for 3 days. Then, mRNA expression levels were evaluated by real-time PCR. Relative GAPDH-normalized expression levels of genes to the control are presented. ## p < 0.01; ### p < 0.001 (vs the vehicle control group); *p < 0.05; **p < 0.01; ***p < 0.001 (vs the BMP-2-treated group)
Mollugin enhances BMP-2-induced mRNA expression of osteogenic BMPs
The effect of mollugin on the mRNA expression levels of BMPs was investigated by real-time PCR analysis. As shown in Fig. 2, BMP-2 significantly induced the mRNA expressions of BMP-2, 4, 6, 7, and 9 in C2C12 cells, and those inductions were dramatically enhanced by mollugin; especially, at 10 μM.
Mollugin induces the phosphorylation of p38 and Smad1/5/8
To elucidate the cellular mechanism of osteo-enhancing mollugin, Western blot analysis was performed to evaluate its effect on the activation of mitogen-activated protein (MAP) kinases and Smad. As shown in Fig. 3a, mollugin induced the phosphorylation of p38 in a dos-dependent manner, but not JNK and ERK, in C2C12 cells in the presence of BMP-2. The phosphorylation of Smad1/5/8 was also induced by mollugin (Fig. 3b). As the biological activity of mollugin has been linked to the regulation of JAK signaling as well as MAP kinases (Zhu et al. 2013), the effect of mollugin on the activity of JAK2 was also evaluated by a kinase activity assay. We found mollugin did not regulate the activity of JAK2 (data not shown).

Mollugin induces the phosphorylation of p38 and Smad1/5/8. The effect of mollugin on the activations of MAP kinases and Smad1/5/8 were evaluated by Western blot analysis. Briefly, C2C12 cells (2 × 105 cells/well) were seeded in a 6-well plate and cultured in GM. After reaching confluence, cells were incubated with DM in the absence or presence of mollugin for 10 min (MAP kinases; a) or 3 days (Smad1/5/8; b). Western blot analysis was then performed as described in the “Materials and methods”
SB202190 inhibits mollugin-enhanced phosphorylation of p38 and Smad1/5/8, and induction of ALP activity
The involvement of the p38–Smad signaling pathway was validated by a pharmacological inhibition study. Consistent with the results shown in Fig. 3, mollugin induced the phosphorylation of both p38 and Smad1/5/8 in C2C12 cells in the presence of BMP-2 (Fig. 4a), but those inductions were prevented, in a dose-dependent manner, by pretreatment with SB202190, a p38 specific inhibitor (Fig. 4a). In C2C12 cells, mollugin-induced ALP activity in the presence of BMP-2 was also significantly prevented by SB202190 in a dose-dependent manner (Fig. 4b).

SB202190 inhibits mollugin-enhanced phosphorylation of p38 and Smad1/5/8, and induction of ALP activity. The inhibitory effects of SB202190 on mollugin-enhanced phosphorylation of p38 and Smad1/5/8 (a), and induction of ALP activity (b) were evaluated in C2C12 cells. Briefly, cells were pre-incubated with SB202190 for 2 h and then cultured with DM in the absence or presence of mollugin for 2 days (for Western blot analysis, a) or 3 days (for ALP activity assay, b). ***p < 0.001 (vs the BMP-2 and mollugin-treated group)
Mollugin enhances the skeletal development of zebrafish
Furthermore, the in vivo effect of mollugin on the skeletal development of zebrafish was investigated, similar to previous studies on several osteo-enhancing compounds (Kim et al. 2012; Baek et al. 2015; Moon et al. 2015). As shown in Fig. 5, mollugin enhanced the development of vertebrae in zebrafish, with the number of alizarin red S-stained vertebrae higher in mollugin-treated zebrafish than the control.

Mollugin enhances the skeletal development of zebrafish. In vivo osteogenic activity of mollugin was investigated in zebrafish as described in the “Materials and methods”. Briefly, zebrafish (5 days after fertilization) were treated with mollugin for 3 days, fixed, and stained with alizarin red S. The appearance of vertebrae (v) is indicated
Discussion
In this study, we found that mollugin enhanced the BMP-2-mediated commitment of C2C12 cells into osteoblasts and enhanced other BMPs at the transcript level. Like BMP-2, the other BMPs investigated in this study also exhibited in vitro and in vivo osteogenic activities (Gitelman et al. 1995; Yamaguchi et al. 1996; Yeh et al. 2002; Li et al. 2003). In addition, the ability of BMP-2 to enhance the gene expression of osteogenic BMPs may play a critical role in bone formation. BMP-2 enhanced the gene expression of other BMPs during osteoblast differentiation (Chen et al. 1997), and the rapid bone formation by the gene transfer of BMP-2 and BMP-7 was also accompanied by increased expression of endogenous BMP-4 (Kawai et al. 2006). These results suggested that the mollugin-enhanced expression of other osteogenic BMPs in the presence of BMP-2 might act synergistically to promote the commitment of C2C12 cells into osteoblasts.
MAP kinases and Smad1/5/8 are essential signaling molecules for BMP-2-induced osteoblast differentiation (Gallea et al. 2001; Lee et al. 2002; Nöth et al. 2003). Here, mollugin enhanced the BMP-2-mediated commitment of C2C12 cells into osteoblasts by accelerating the activation of p38 and Smad1/5/8. In addition, a pharmacological inhibition study confirmed the involvement of the p38–Smad1/5/8 signaling axis in mollugin’s enhancement of the osteogenic activity of BMP-2. The p38 inhibitor, SB202190, hindered the mollugin-enhanced phosphorylation of p38 and Smad1/5/8, and induction of ALP activity in C2C12 cells. The activation of p38 controls the expression of ALP during the osteoblastic differentiation of osteoblast-like cells or stromal cells (Suzuki et al. 2002; Nishikawa et al. 2014). BMP-2 can trigger the osteogenic signaling through the action of Smad, and the phosphorylation of Smad then induces the expression of genes related to osteoblast differentiation (Nishimura et al. 1998; Yamachika et al. 2009).
The in vitro osteo-enhancing activity of mollugin further led us to investigate its in vivo effect on the skeletal development of zebrafish. The mammalian bone forming pathway has been suggested to be conserved during the skeletal development of zebrafish (Li et al. 2009), and zebrafish are successful models for identifying bone-formimg agents (Du et al. 2001; Fleming et al. 2005). When zebrafish embryos were treated with mollugin, it promoted the rate of vertebral development.
In summary, mollugin has the potential to enhance the osteogenic activity of BMP-2 in C2C12 cells by accelerating the activation of the p38–Smad1/5/8 signaling axis. In addition, mollugin promoted skeletal development in zebrafish. When considering the anti-osteoclastic activity of mollugin (Baek et al. 2015), its dual action in inhibiting bone resorption and stimulating bone formation might be beneficial in treating bone-related disorders. Moreover, the combination of BMP-2 with osteogenic small molecules could minimize the limitations associated with BMP-2. Furthermore, since chemical biologists in stem cell research are trying to identify bioactive small molecules to direct stem cells to a lineage-specific patterns (differentiation), mollugin might lead to the development of effective stem cell stimulants for tissue repair, especially bone regeneration and fracture healing (Hong et al. 2011; Choi and Nam 2012).
References
Alt V, Donell ST, Chhabra A, Bentley A, Eicher A, Schnettler R (2009) A health economic analysis of the use of rhBMP-2 in Gustilo-Anderson grade III open tibial fractures for the UK, Germany, and France. Injury 40:1269–1275
Baek JM, Kim JY, Jung Y, Moon SH, Choi MK, Kim SH, Lee MS, Kim I, Oh J (2015) Mollugin from Rubea cordifolia suppresses receptor activator of nuclear factor-κB ligand-induced osteoclastogenesis and bone resorbing activity in vitro and prevents lipopolysaccharide-induced bone loss in vivo. Phytomedicine 22:27–35
Benglis D, Wang MY, Levi AD (2008) A comprehensive review of the safety profile of bone morphogenetic protein in spine surgery. Neurosurgery 62:ONS423–ONS431
Cao Y, Wang C, Zhang X, Xing G, Lu K, Gu Y, He F, Zhang L (2014) Selective small molecule compounds increase BMP-2 responsiveness by inhibiting Smurf1-mediated Smad1/5 degradation. Sci Rep 14:4965
Chen D, Harris MA, Rossini G, Dunstan CR, Dallas SL, Feng JQ, Mundy GR, Harris SE (1997) Bone morphogenetic protein 2 (BMP-2) enhances BMP-3, BMP-4, and bone cell differentiation marker gene expression during the induction of mineralized bone matrix formation in cultures of fetal rat calvarial osteoblasts. Calcif Tissue Int 60:283–290
Cheng H, Jiang W, Phillips FM, Haydon RC, Peng Y, Zhou L, Luu HH, An N, Breyer B, Vanichakarn P, Szatkowski JP, Park JY, He TC (2003) Osteogenic activity of the fourteen types of human bone morphogenetic proteins (BMPs). J Bone Joint Surg Am 8:1544–1552
Choi Y, Nam TG (2012) Chemical biology in stem cell research. Arch Pharm Res 35:281–297
Do MT, Hwang YP, Kim HG, Na M, Jeong HG (2013) Mollugin inhibits proliferation and induces apoptosis by suppressing fatty acid synthase in HER2-overexpressing cancer cells. J Cell Physiol 228:1087–1097
Du SJ, Frenkel V, Kindschi G, Zohar Y (2001) Visualizing normal and defective bone development in zebrafish embryos using the fluorescent chromophore calcein. Dev Biol 238:239–246
Fan J, Guo M, Im CS, Pi-Anfruns J, Cui ZK, Kim S, Wu B, Aghaloo T, Lee M (2016) Enhanced mandibular bone repair by combined treatment of bone morphogenetic protein 2 (BMP-2) and small molecule phenamil. Tissue Eng Part A 22:195–207
Fleming A, Sato M, Goldsmith P (2005) High-throughput in vivo screening for bone anabolic compounds with zebrafish. J Biomol Screen 10:823–831
Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S, Kawai S, Faucheu C, Huet L, Baron R, Roman-Roman S (2001) Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone 28:491–498
Gitelman SE, Kirk M, Ye JQ, Filvaroff EH, Kahn AJ, Derynck R (1995) Vgr-1/BMP-6 induces osteoblastic differentiation of pluripotential mesenchymal cells. Cell Growth Differ 6:827–836
Ho LK, Don MJ, Chen HC, Yeh SF, Chen JM (1996) Inhibition of hepatitis B surface antigen secretion on human hepatoma cells. Components from Rubia cordifolia. J Nat Prod 59:330–333
Hong HS, Kim DY, Yoon KJ, Son Y (2011) A new paradigm for stem cell therapy: substance-P as a stem cell-stimulating agent. Arch Pharm Res 34:2003–2006
James AW, LaChaud G, Shen J, Asatrian G, Nguyen V, Zhang X, Ting K, Soo C (2016) A review of the clinical side effects of bone morphogenetic protein-2. Tissue Eng Part B 22:284–297
Jeong GS, Lee DS, Kim DC, Jahng Y, Son JK, Lee SH, Kim YC (2011) Neuroprotective and anti-inflammatory effects of mollugin via up-regulation of heme oxygenase-1 in mouse hippocampal and microglial cells. Eur J Pharmacol 654:226–234
Jun do Y, Han CR, Choi MS, Bae MA, Woo MH, Kim YH (2011) Effect of mollugin on apoptosis and adipogenesis of 3T3-L1 preadipocytes. Phytother Res 25:724–731
Kawai M, Bessho K, Maruyama H, Miyazaki J, Yamamoto T (2006) Simultaneous gene transfer of bone morphogenetic protein (BMP) -2 and BMP-7 by in vivo electroporation induces rapid bone formation and BMP-4 expression. BMC Musculoskelet Disord 7:62
Kawasaki Y, Goda Y, Yoshihira K (1992) The mutagenic constituents of Rubia tinctorum. Chem Pharm Bull (Tokyo) 40:1504–1509
Kim HJ, Kim SH (2010) Tanshinone IIA enhances BMP-2-stimulated commitment of C2C12 cells into osteoblasts via p38 activation. Amino Acids 39:1217–1226
Kim SN, Kim MH, Min YK, Kim SH (2008) Licochalcone A inhibits the formation and bone resorptive activity of osteoclasts. Cell Biol Int 32:1064–1072
Kim KJ, Lee JS, Kwak MK, Choi HG, Yong CS, Kim JA, Lee YR, Lyoo WS, Park YJ (2009) Anti-inflammatory action of mollugin and its synthetic derivatives in HT-29 human colonic epithelial cells is mediated through inhibition of NF-κB activation. Eur J Pharmacol 622:52–57
Kim SN, Bae SJ, Kwak HB, Min YK, Jung SH, Kim CH, Kim SH (2012) In vitro and in vivo osteogenic activity of licochalcone A. Amino Acids 42:1455–1465
Kwak HB, Yang D, Ha H, Lee JH, Kim HN, Woo ER, Lee S, Kim HH, Lee ZH (2006) Tanshinone IIA inhibits osteoclast differentiation through down-regulation of c-Fos and NFATc1. Exp Mol Med 38:256–264
Lee KS, Hong SH, Bae SC (2002) Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene 21:7156–7163
Lee YR, Wang X, Kim YM, Shim JJ, Kim BN, Han DH (2007) Concise synthesis of biologically interesting mollugins and its analogues. Bull Korean Chem Soc 28:1735–1738
Lee YM, Auh QS, Lee DW, Kim JY, Jung HJ, Lee SH, Kim EC (2013) Involvement of Nrf2-mediated upregulation of heme oxygenase-1 in mollugin-induced growth inhibition and apoptosis in human oral cancer cells. Biomed Res Int 2013:210604
Li JZ, Li H, Sasaki T, Holman D, Beres B, Dumont RJ, Pittman DD, Hankins GR, Helm GA (2003) Osteogenic potential of five different recombinant human bone morphogenetic protein adenoviral vectors in the rat. Gene Ther 10:1735–1743
Li N, Felber K, Elks P, Croucher P, Roehl HH (2009) Tracking gene expression during zebrafish osteoblast differentiation. Dev Dyn 238:459–466
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408
Lrad TW, Einhorn TA (2009) Bone morphogenetic proteins in orthopaedic surgery. Cytokine Growth Factor Rev 20:481–488
McKay WF, Peckham SM, Badura JM (2007) A comprehensive clinical review of recombinant human bone morphogenetic protein-2 (INFUSE bone graft). Int Orthop 31:729–734
Mondal S, Bandyopadhyay S, Ghosh MK, Mukhopadhyay S, Roy S, Mandal C (2012) Natural products: promising resources for cancer drug discovery. Anticancer Agents Med Chem 12:49–75
Moon SH, Choi SW, Park SJ, Ryu SY, Hwang KS, Kim CH, Kim SH (2015) In vitro and in vivo bone-forming activity of Saururus chinensis extract. Phytother Res 7:1073–1080
Nishikawa R, Anada T, Ishiko-Uzuka R, Suzuki O (2014) Osteoblastic differentiation of stromal ST-2 cells from octacalcium phosphate exposure via p38 signaling pathway. Dent Mater J 33:242–251
Nishimura R, Kato Y, Chen D, Harris SE, Mundy GR, Yoneda T (1998) Smad5 and DPC4 are key molecules in mediating BMP-2-induced osteoblastic differentiation of the pluripotent mesenchymal precursor cell line C2C12. J Biol Chem 273:1872–1879
Nöth U, Tuli R, Seghatoleslami R, Howard M, Shah A, Hall DJ, Hickok NJ, Tuan RS (2003) Activation of p38 and Smads mediates BMP-2 effects on human trabecular bone-derived osteoblasts. Exp Cell Res 291:201–211
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386
Suzuki A, Guicheux J, Palmer G, Miura Y, Oiso Y, Bonjour JP, Caverzasio J (2002) Evidence for a role of p38 MAP kinase in expression of alkaline phosphatase during osteoblastic cell differentiation. Bone 30:91–98
Weaver CM, Alekel DL, Ward WE, Ronis MJ (2012) Flavonoid intake and bone health. J Nutr Gerontol Geriatr 31:239–253
Yamachika E, Tsujigiwa H, Shirasu N, Ueno T, Sakata Y, Fukunaga J, Mizukawa N, Yamada M, Sugahara T (2009) Immobilized recombinant human bone morphogenetic protein-2 enhances the phosphorylation of receptor-activated Smads. J Biomed Mater Res A 88:599–607
Yamaguchi A, Ishizuya T, Kintou N, Wada Y, Katagiri T, Wozney JM, Rosen V, Yoshiki S (1996) Effects of BMP-2, BMP-4, and BMP-6 on osteoblastic differentiation of bone marrow-derived stromal cell lines, ST2 and MC3T3-G2/PA6. Biochem Biophys Res Commun 220:366–371
Yeh LC, Tsai AD, Lee JC (2002) Osteogenic protein-1 (OP-1, BMP-7) induces osteoblastic cell differentiation of the pluripotent mesenchymal cell line C2C12. J Cell Biochem 87:292–304
Zhu ZG, Jin H, Yu PJ, Tian YX, Zhang JJ, Wu SG (2013) Mollugin inhibits the inflammatory response in lipopolysaccharide-stimulated RAW264.7 macrophages by blocking the Janus kinase-signal transducers and activators of transcription signaling pathway. Biol Pharm Bull 36:399–406
Acknowledgements
This work was supported by a project Grant (KK1703-F02) from the Korea Research Institute of Chemical Technology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have declared no conflict of interest.
Rights and permissions
About this article

Cite this article
Moon, SH., Kim, I. & Kim, S.H. Mollugin enhances the osteogenic action of BMP-2 via the p38–Smad signaling pathway. Arch. Pharm. Res. 40, 1328–1335 (2017). https://doi.org/10.1007/s12272-017-0964-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-017-0964-4