
Abstract
A fast and efficient microwave-assisted solid phase peptide synthesis (MW-SPPS) of a 51mer peptide, the main heparin-binding site (60–110) of human pleiotrophin (hPTN), using 2-chlorotrityl chloride resin (CLTR-Cl) following the 9-fluorenylmethyloxycarbonyl/tert-butyl (Fmoc/tBu) methodology and with the standard N,N′-diisopropylcarbodiimide/1-hydroxybenzotriazole (DIC/HOBt) coupling reagents, is described. An MW-SPPS protocol was for the first time successfully applied to the acid labile CLTR-Cl for the faster synthesis of long peptides (51mer peptide) and with an enhanced purity in comparison to conventional SPPS protocols. The synthesis of such long peptides is not trivial and it is generally achieved by recombinant techniques. The desired linear peptide was obtained in only 30 h of total processing time and in 51% crude yield, in which 60% was the purified product obtained with 99.4% purity. The synthesized peptide was purified by reversed phase high performance liquid chromatography (RP-HPLC) and identified by electrospray ionization mass spectrometry (ESI-MS). Then, the regioselective formation of the two disulfide bridges of hPTN 60–110 was successfully achieved by a two-step procedure, involving an oxidative folding step in dimethylsulfoxide (DMSO) to form the Cys77–Cys109 bond, followed by iodine oxidation to form the Cys67–Cys99 bond.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pleiotrophin (PTN) is a heparin-binding growth factor with diverse biological activities, the most studied being those related to the nervous system, tumor growth and angiogenesis. PTN actions are mediated by its interaction with diverse receptors, such as N-syndecan, anaplastic lymphoma kinase, receptor protein tyrosine phosphatase β/ζ and integrin ανβ3 (Mikelis et al. 2007; Papadimitriou et al. 2009). It has been hypothesized for several years that different regions of the molecule exert distinct functions. For example, the C-terminal lysine-rich domain of PTN (amino acids 112–136) is not involved in neurite outgrowth activity, but it seems to play a key role in the mitogenic and tumor formation activities of PTN (Bernard-Pierrot et al. 2001; Ducès et al. 2008). The two thrombospondin type 1 repeat (TSR-1) motifs have been long considered responsible for the interaction of PTN with heparin (Kilpelainen et al. 2000), an interaction associated with many of the biological activities of PTN, among which are the interaction and inhibition of the angiogenic activities of vascular endothelial growth factor (Héroult et al. 2004; Polykratis et al. 2004). More recent data suggest the involvement of only the carboxyl terminal TSR-1 motif in heparin binding (Hamma-Kourbali et al. 2008; Ori et al. 2009), and the mitogenic, transforming and angiogenic activities of PTN in vitro and in vivo in nude mice (Hamma-Kourbali et al. 2008). It is interesting, therefore, to determine which regions of PTN are responsible for its diverse functions, in order to identify the molecular mechanisms involved and to identify possible therapeutic targets or/and agents.
Full length human PTN (hPTN) (Fig. 1), is being produced as a recombinant protein (Papadimitriou et al. 2000; Kilpelainen et al. 2000) and is commercially available from several different sources. It has not been made possible, however, to produce high amounts of pure protein, necessary for structural studies, mainly due to solubility problems (Seddon et al. 1994). Synthesis of recombinant hPTN peptides has become possible; however, GST tag was used in all cases (Heroult et al. 2004; Polykratis et al. 2004; Raulo et al. 2005). We have tried to produce high amounts of hPTN or hPTN peptides by using GST or His tag; however, removal of the tag leads to very low yields, insufficient to perform structural studies (Mikelis et al., unpublished data). An alternative approach in order to get high amounts of hPTN or hPTN peptides would be peptide synthesis. So far, there has been one attempt by using liquid phase peptide synthesis with the Boc/Bzl orthogonal protection strategy and fragment condensation techniques (Inui et al. 2000). The main drawbacks of this technique are the usage of the highly toxic HF and the low yield of the final product due to the insolubility problem, which increases with the size of the peptide. In order to overcome this problem, solvent additives in a coupling step have to be used, e.g. TFE or a mixture of CHCl3/TFE (Yamashiro et al. 1976; Felix et al. 1985), an approach which can be effective, but is troublesome and expensive. Therefore, the development of efficient direct methods for the solid-phase synthesis of high amounts of hPTN or hPTN peptides are of great importance for both structural and structure–function studies.

In peptide synthesis, microwave irradiation has been applied as a fast and efficient way to synthesize large peptide sequences with high yields and low racemization (Palasek et al. 2007; Bacsa et al. 2010). Fmoc deprotection and coupling reactions are not only accelerated due to increased temperature, but also due to the alternating electric field of the microwave (Hayes 2002), to which the polar backbone of the peptide continuously tries to align. During peptide synthesis this can lead to decreased steric hindrance and prevention of chain aggregation, allowing for easier access of the reagents to the solid phase reaction matrix and reducing the Fmoc deprotection and coupling reaction times.
An advance in peptide chemistry has been the development of microwave-enhanced solid phase peptide synthesis (MW-SPPS) (Hayes 2002; Kappe 2004; Caddick and Fitzmaurice 2009; Sabatino and Papini 2008; Galanis et al. 2009; Heggemann et al. 2010) which in combination with CLTR-Cl (Barlos et al. 1989) allows the fast stepwise synthesis of large and difficult peptide sequences. Up to now, reports regarding MW-SPPS utilizing the acid labile CLTR-Cl for the synthesis of long peptides, with more than 20aa, do not exist. Herein, we report the fast and efficient, microwave-assisted step by step synthesis of the main heparin-binding site of PTN that corresponds to amino acids 60–110 (hPTN60–110), a 51mer peptide which contains two disulfide bonds, one between the 67–99 residues and one between the 77–109 residues (Hulmes et al. 1993). We employed the Fmoc/tBu orthogonal protection strategy, which utilizes the standard and inexpensive DIC/HOBt coupling reagents, followed by the regioselective formation of the two disulfide bonds in solution.
The Fmoc/tBu orthogonal protection strategy (Sewald and Jakubke 2002; Kates and Albericio 2000; Chan and White 2000) has been applied in the synthesis of a series of linear and cyclic analogs of important peptides, such as angiotensin implicated in hypertension (Matsoukas et al. 1993, 1994), myelin peptides implicated in multiple sclerosis (Spyranti et al. 2007; Friligou et al. 2008; Katsara et al. 2009) and thrombin receptor activating peptides implicated in angiogenesis and cancer (Matsoukas et al. 1996; Alexopoulos et al. 2004).
Materials and methods
2-Chlorotrityl chloride polystyrene resin (1% DVB, 200–400 mesh), Fmoc-protected-amino acids and piperidine were purchased from CBL-Patras (Patras, Greece). Solvents and other reagents were purchased from Merck (Darmstadt, Germany), Sigma-Aldrich (Steinheim, Germany), Fluka (Buchs, Switzerland) and Panreac (Barcelona, Spain).
The microwave synthesis was performed on a LibertyTM Microwave Peptide Synthesizer (CEM Corporation, Matthews, NC), an additional module of DiscoverTM (CEM Corporation, Matthews, NC) using a frequency of 2,450 MHz.
The determination of resin loading capacity and the quantification of thiol groups were achieved spectrophotometrically using a UV/Visible Spectrophotometer Cary 50 CONC.
ESI-MS spectra were recorded on a Waters Micromass ZQ 4000 mass detector (positive mode), controlled by the MassLynx 4.1 software. Cone voltage was set at 30 V and scan time at 1 s, with interscan delay at 0.1 s.
Peptide synthesis
First amino acid loading
The linear peptide was prepared on CLTR-Cl (0.100 g, 0.7 mmol Cl−/g resin) (Fig. 2). The first Nα-Fmoc (9-fluorenylmethyloxycarbonyl) protected amino acid, Fmoc-Gly-OH (0.8 equiv.), was coupled (esterified) to the resin in the presence of DIPEA (4.5 equiv.) in DCM for 1.5 h at RT. The resin was washed successively with DCM (3 × 5 mL), DMF (3 × 5 mL) and i-PrOH (3 × 5 mL) and dried overnight under vacuum. The loading of the resin (0.30 mmol/g resin) was determined spectrophotometrically (301 nm, ε: 7,800 M−1 cm−1) from the amount of the adduct dibenzofulvene-piperidine formed after treatment of the Fmoc-Gly-resin with piperidine/DCM/MeOH. The remaining active site of the resin was capped using a mixture of DCM/MeOH/DIPEA (85:10:5) for 15 min at RT. The Fmoc-Gly-resin was subsequently filtered and washed with DCM (1 × 5 mL), DMF (3 × 5 mL), i-PrOH (2 × 5 mL) and n-hexane (1 × 5 mL) and dried overnight under vacuum.

Solid phase peptide synthesis of linear [Cys(Acm)67,99]-hPTN60–110 (1)
Solid phase peptide synthesis of linear [Cys(Acm)67,99]-hPTN60–110 (1)
The next two amino acids were coupled manually (2.5 equiv. of the appropriate Fmoc-amino acid) in the presence of DIC (2.75 equiv.) and HOBt followed by Fmoc deprotection with piperidine (20% in DMF, 2 × 20 min). The completeness of each coupling and deprotection step was verified by the Kaiser test and thin layer chromatography (TLC) using 1-BuOH/AcOH/H2O (4:1:1) and Tol/MeOH/AcOH (7:1.5:1.5) as solvent systems. The remaining protected peptide chain was assembled under microwave irradiation following the Fmoc/tBu methodology. Fmoc deprotection was performed with a solution of 20% piperidine in DMF. All coupling reactions were performed in the presence of a 5-fold molar excess of 0.2 M Fmoc-protected amino acids dissolved in DMF with a 10-fold molar excess of 0.5 M HOBt/DIC dissolved in DMF as activators (Table 1).
Amino acid side-chain protection was as follows: tBu for Asp, Glu, Tyr, Thr, Ser; Pbf for Arg; Boc for Lys and Trp; Trt for Asn, Gln and His. Cys at positions 67 and 99 was incorporated as S-Acm derivative, while Cys at positions 77 and 109 was incorporated as S-Trt derivative.
The synthesized protected peptide on the resin was dried under vacuum and then cleaved with the splitting solution DCM/TFE/AcOH (7:2:1) for 1 h at RT. The mixture was filtered, the solvent was removed on a rotary evaporator and the obtained oily product was precipitated by the addition of cold diethyl ether as an amorphous white solid (0.228 g). The deprotection of the linear protected peptide (1a) was achieved using TFA/EDT/H2O solution (90:5:5 v/v/v) for 5 h at RT. The solvent was partially evaporated and the crude product (1) was precipitated with cold diethyl ether and collected by filtration (0.150 g).
Purification and characterization of linear [Cys(Acm)67,99]-hPTN60–110 (1)
The crude peptide product (1) was analyzed by analytical HPLC (Waters 600 solvent delivery system, combined with a Waters 996 photodiode array detector) using a Nucleosil C-18 column (5 μm, 250 × 4 mm) at 214 and 254 nm. Separation was achieved by gradient elution of 5–100% solvent B (solvent A = 0.08% TFA in H2O; solvent B = 0.08% TFA in ACN) over 30 min at a flow rate of 1 mL/min (conditions I).
The purification was carried out on a semi-preparative RP-HPLC (Waters 600 solvent delivery system, combined with a Waters 996 photodiode array detector) using a Nucleosil C-18 column (7 μm, 250 × 10 mm) at 214 and 254 nm. Separation was achieved by gradient elution of 20–50% solvent B (solvent A = 0.08% TFA in H2O; solvent B = 0.08% TFA in ACN) over 45 min at a flow rate of 3 mL/min (conditions II).
The final peptide product (1) was lyophilized [yield: 0.09 g, (60%), HPLC purity: 99.4%] and it was analyzed by analytical HPLC (Waters Alliance 2695 Separations Module combined with Waters 2996 photodiode array detector) using a C-8 Purospher column (5 μm, 250 × 4 mm) at 214 and 254 nm. Separation was achieved by gradient elution of 5–100% solvent B (solvent A = 0.08% TFA in H2O; solvent B = 0.08% TFA in ACN) over 30 min at a flow rate of 1 mL/min (conditions III). The final product was identified by ESI-MS: M calc. = 5,831.64 Da, M found = 5,832.05 Da.
Oxidative formation of the first disulfide bond by dimethyl sulfoxide
For the formation of the Cys77, Cys109-disulfide bridge, the pure peptide (1) (10 mg, 1.7 μmol) was dissolved (0.5 mg/mL) in a DMSO/0.1 M AcONH4 (1:4) mixture (pH 6.0) and the solution were stirred at RT for 24 h. The solution was subjected to semi-preparative HPLC purification (conditions II) to yield, after lyophilization, the monocyclic [Cys(Acm)67,99]-hPTN60–110 (2) [yield: 6.9 mg (69%), HPLC purity: 99.5%]. The product was identified by ESI-MS: M calc. = 5,829.63 Da, M found = 5,829.04 Da. The completeness of the oxidation was determined by measuring residual-free thiols (<2%) on 250 μl aliquot mixed with 2.5 mL of reaction buffer (0.1 M sodium phosphate, pH 8.0, containing 1 mM EDTA) and 50 μl of Ellman’s Reagent solution (4 mg Ellman’s Reagent in 1 mL of reaction buffer). The absorbance was read at 412 nm (ε: 141,50 M−1 cm−1) (Riddles et al. 1979).
Deprotection-oxidation of Cys(Acm) with iodine
A 0.35 mM solution of monocyclic [Cys(Acm)67,99]-hPTN60–110 (2) (5 mg, 0.86 μmol) in AcOH/H2O (4:1) was added dropwise into a 3.5 mM solution of iodine in AcOH/H2O (4:1), corresponding to 5 equiv./S-Acm function and the solution was stirred for a further 10 min. The reaction was quenched by adding ascorbic acid until the brown color of iodine disappeared. Excess iodine was extracted twice with CHCl3. The aqueous phase was lyophilized to give the final oxidized peptide, followed by purification on semi-preparative HPLC (conditions II). The final product (3) [yield: 2.15 mg (43%), HPLC purity: 98.1%] was further identified by ESI-MS: M calc. = 5,685.46 Da, M found = 5,685.08 Da. The completeness of the reaction was determined by measuring residual-free thiols with Ellman’s Reagent as described above (<1%).
Results and discussion
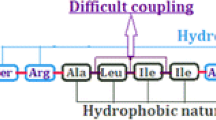
Herein, we report for the first time the usage of the acid-labile CLTR-Cl for the step by step MW-SPPS of a large (51mer) peptide utilizing the inexpensive DIC/HOBt coupling reagents. We combined the benefits of CLTR-Cl, which presents high swelling properties in DMF, the main solvent used in MW-SPPS, with the benefits of microwave irradiation, leading to the rapid and efficient synthesis of hPTN60–110. As a number of reports have highlighted during the past few years (Kent et al. 1992; Rizzolo et al. 2007), also in our occasion, the step by step synthesis of hPTN60–110 using conventional SPPS protocols would present difficulties in terms of slow and incomplete couplings and Fmoc-removal reactions, due to chain aggregation, leading to low yields (<20%) because of byproducts, deletion peptides and truncated sequences.
Since the 60–110 domain of hPTN contains two disulfide bridges (Cys67–Cys99 and Cys77–Cys109), in order to achieve the regioselective folding of the peptide, we decided to follow a two-step strategy (Vasileiou et al. 2010; Góngora-Benítez et al. 2010; Galanis et al. 2008), where the first disulfide bond is formed and then the second one in a subsequent step (Fig. 3). Cys77,109 were protected with the Trt group which generates the free thiol upon deprotection with TFA, while Cys67,99 were protected with the Acm group, which is acid- and base-stable.

Synthetic procedure of regioselective formation of disulfide bonds of hPTN60–110
Microwave-enhanced solid-phase peptide synthesis of linear [Cys(Acm)67,99]-hPTN60–110 (1)
The linear protected peptide (1a) was synthesized on CLTR-Cl using the standard Fmoc/tBu methodology. In order to avoid (1) premature cleavage of the first di- or three-peptide from the resin due to its sensitivity, and (2) insufficient couplings because of the steric hindrance of the 2-chlorotrityl linker, the first three amino acids were incorporated by the conventional SPPS methodology. The chain elongation of the rest of the peptide was performed using automatic MW-SPPS. In both conventional and MW-SPPS, DIC/HOBt was used as coupling reagent. The Fmoc-removal was performed by treatment with 20% piperidine in DMF. Coupling reactions and the quantitative removal of the Fmoc-group, during conventional synthesis, were checked with the Kaiser test and TLC.
Initially, we tried to perform the MW-SPPS with the standard conditions, in terms of power and temperature, used for Wang type resins, but we obtained a low yield (<40%) of the protected peptide on resin. Probably, this is due to the high sensitivity of CLTR, which may not be stable at elevated temperatures and after performing repetitive cycles of microwave. Since microwave irradiation generates higher temperature, His could be prone to racemization, because of the π-nitrogen which is closer to the α-carbon and sufficiently basic to abstract a proton. As a consequence, it was used a particular coupling cycle, composed of two steps, at 50°C, to introduce this residue in the peptide sequence (Palasek et al. 2007). As with histidine, cysteine requires special attention. Cysteine racemization has been attributed to base-catalyzed α-carbon proton abstraction during coupling. The level of racemization can be decreased by using weaker bases and avoiding preactivation in phosphonium or aminium salt-mediated coupling protocols (Han et al. 1997). Additionally, in order to overcome increases in racemization from excess microwave energy, coupling temperature was reduced to 50°C (Palasek et al. 2007). Once both histidine and cysteine are incorporated, they do not show any further increase in racemization during chain elongation. Moreover, it is well known that Fmoc-Arg(Pbf)-OH, during activation step at high temperatures, is prone to formation of δ-lactam with an approximately 80% conversion. In order to avoid this side reaction, it was performed a particular coupling cycle, composed of two steps, at low temperature (White et al. 2005). Thus, also taking into consideration the hydrophobicity of CLTR in comparison to the other Wang type resins, we decided to adopt modified MW-SPPS conditions by decreasing the temperature from 75°C to 60°C and increasing a little the Watts (from 25 to 34 W, ×1.36) in order to obtain faster the desired temperature (Table 1). Moreover, three different types of microwave synthetic conditions were used with low yield (<40%) in synthetic product as follows: coupling cycles at 75°C (25 W), 60°C (30 W) and 50°C (35 W); initial deprotection cycles at 75°C (25 W), 60°C (35 W) and 50°C (35 W); deprotection cycles at 75°C (35 W), 60°C (62 W) and 50°C (65 W).
One well-known side product of SPPS using Cys(Acm) is the formation of piperidyl alanine, specially in the case of C-terminal Cys, which in the Fmoc/tBu methodology can undergo β-elimination followed by piperidine addition to give piperidyl alanine (Isidro-Llobet et al. 2009). Although it is expected that this side product will be found in higher amounts in products obtained at higher temperatures, in our case was not observed. One possible explanation could be the ability of microwave to accelerate the Fmoc-removal (initial deprotection time: 30 s, Table 1).
Cleavage from resin: deprotection and purification
The protected peptide was cleaved from CLTR by treatment of the peptide-resin ester with DCM/TFE/AcOH (7:2:1) for 1 h at RT. The protected peptide (1a) was obtained in 80% yield.
We treated the protected peptide (1a) with various mixtures in order to choose the most suited deprotection cocktail (data not shown). We found that treatment of the protected peptide with cleavage mixtures containing TFA/EDT/H2O (90:5:5) for 5 h at RT gave the best results in terms of purity.
The crude partially deprotected peptide (with two Acm groups) (1) was subsequently analyzed by analytical RP-HPLC (conditions I) (Fig. 4), purified by semi-preparative RP-HPLC (conditions II) and verified with ESI-MS (Fig. 5b), to yield after lyophilization the linear [Cys(Acm)67,99]-hPTN60–110 (1) in 99.4% purity, as determined by RP-HPLC analysis (conditions III) (Fig. 5a).

Analytical RP-HPLC (conditions I) of crude linear [Cys(Acm)67−99]-hPTN60–110 (1)

a Analytical RP-HPLC (conditions III) of purified linear [Cys(Acm)67−99]-hPTN60–110 (1). b ESI-MS of the purified linear [Cys(Acm)67−99]-hPTN60–110 (1) after lyophilization
Synthesis of monocyclic [Cys(Acm)67,99]-hPTN60–110 (2)
The purified linear [Cys(Acm)67,99]-hPTN60–110 (1) was dissolved in a DMSO/0.1 M AcONH4 (20:80) mixture (pH 6.0) (Albericio et al. 1991; Tam et al. 1991). The folding reaction was completed after 24 h at RT, as verified by analytical HPLC and by measuring spectrophotometrically residual-free thiols. The reaction mixture was directly subjected to semi-preparative HPLC purification (conditions II) to yield, after lyophilization, monocyclic [Cys(Acm)67,99]-hPTN60–110 (2) in 99.5% purity. The final product was further analyzed by RP-HPLC (conditions III) and identified by ESI-MS (Fig. 6a, b). The measured mass of monocyclic [Cys(Acm)67,99]-hPTN60–110 (2) was consistent with the formation of one disulfide bond (M calc. = 5,829.63 Da; M found = 5,829.04 Da). The formation of the first disulfide bond, i.e. 2.6 Da decrease, was corresponding to the loss of two protons.

a Analytical RP-HPLC (conditions III) of purified monocyclic [Cys(Acm)67−99]-hPTN60–110 (2). b ESI-MS of the purified monocyclic [Cys(Acm)67−99]-hPTN60–110 (2)
Synthesis of bicyclic hPTN60–110 (3)
For the simultaneous deprotection-oxidation of the two Cys(Acm)67,99 residues, monocyclic [Cys(Acm)67,99]-hPTN60–110 (2) was dissolved in a AcOH/H2O (4:1) mixture and was added dropwise into a solution of iodine (5 equiv./S-Acm) in AcOH/H2O (4:1) (Kamber 1973). The formation of the disulfide bond was fast, completed in 10 min, as revealed by spectrophotometric evaluation of residual-free thiols.
The crude bicyclic hPTN60–110 (3) was purified by semi-preparative RP-HPLC (conditions II) to yield, after lyophilization, bicyclic hPTN60–110 (3) in 98.1% purity. The final product was further analyzed by RP-HPLC (conditions III) and identified by ESI-MS (Fig. 7a, b). The actual mass of the fully oxidized peptide (M found = 5,685.08 Da) was consistent with the calculated mass (M calc. = 5,685.46 Da). Table 2 summarizes the analytical data of the final peptides.

a Analytical RP-HPLC (conditions III) of purified bicyclic hPTN60–110 (3). b ESI-MS of the purified bicyclic hPTN60–110 (3)
RP-HPLC analysis revealed that the main byproduct of the iodine oxidation was due to the oxidative degradation of Trp (a mass difference corresponding to 32 Da) (Nagata et al. 1992; Finley et al. 1998). Also, when we increased the quantity of iodine (from 5 equiv. to 50 equiv./S-Acm) in order to minimize the reaction time, even when the mixture was left to react for no more than 5 min, the only product obtained was the one corresponding to the oxidatively degradated Trp. On the contrary, in both occasions, no byproduct was observed corresponding to the intramolecular S → N or S → O migration of Acm group to the side-chains of Ser, Thr or Gln (Lamthanh et al. 1993, 1995).
Both folding procedures took place in a dilute solution of the peptide in order to prevent the formation of aggregates and polymers. Regarding iodine oxidation reaction, it is usually carried out in mixtures of aq. MeOH or aq. AcOH (Kamber et al. 1980). The choice of the solvent is sequence dependent. Reactions are fastest in aq. MeOH, but since our peptide sequence contained Tyr, His and Trp, we preferred to use aq. AcOH as it limits iodination of these sensitive residues (Lamthanh et al. 1993), as well as the formation of tryptophan thioesters (Sieber 1977).
Nagata et al. (1992) have suggested that the oxidative degradation of Trp residue might be suppressed due to the protonation of the indole nucleus. They found that degradation of Trp is suppressed when the iodine oxidation of Trp-containing peptides is performed in 95% aq. AcOH in the presence of HCl (13 M equivalents to Trp). Thus, we initially tried to perform iodine oxidation in 95% aq. AcOH, but we had solubility problems of the peptide and so we performed it in 90% aq. AcOH in the presence of HCl. After 20 min we performed an RP-HPLC analysis which revealed that the side reaction of Trp degradation was eliminated but the reaction rate slowed down since, apart from the desired bicyclic peptide, we could still find a peak corresponding to the monocyclic product that had not reacted (data not shown).
Conclusions
In conclusion, we have developed an improved synthetic protocol for the MW-SPPS utilizing the acid labile CLTR-Cl and we have successfully applied it for the synthesis of a 51mer peptide, the main heparin-binding site of PTN (60–110). The desired linear peptide was obtained in only 30 h of total processing time in high yield (60% crude yield) and purity. The regioselective formation of the two disulfide bonds was achieved by a two-step strategy, using the Trt and Acm orthogonal protective groups for Cys. We also verified that iodine oxidation performed in an acidic solvent suppresses the degradation of Trp.
Abbreviations
- Acm:
-
Acetamidomethyl
- ACN:
-
Acetonitrile
- AcOH:
-
Acetic acid
- Boc:
-
tert-Butyloxybarbonyl
- 1-BuOH:
-
N-butanol
- Bzl:
-
Benzyl
- CLTR-Cl:
-
2-Chlorotrityl chloride
- DCM:
-
Dichloromethane
- DIC:
-
N,N′-diisopropylcarbodiimide
- DIPEA:
-
Diisopropylethyamine
- DMF:
-
N,N′-dimethylformamide
- DMSO:
-
Dimethylsulfoxide
- EDT:
-
1,2-Ethanedithiol
- ESI-MS:
-
Electrospray ionization mass spectrometry
- Fmoc:
-
9-Fluorenylmethyloxycarbonyl
- HOBt:
-
1-Hydroxybenzotriazole
- hPTN:
-
Human pleiotrophin
- i-PrOH:
-
Isopropanol
- MeOH:
-
Methanol
- MW-SPPS:
-
Microwave-assisted solid-phase peptide synthesis
- PTN:
-
Pleiotrophin
- rhPTN:
-
Recombinant hPTN
- tBu:
-
tert-Butyl
- RP-HPLC:
-
Reversed phase high performance liquid chromatography
- TFA:
-
Trifluoroacetic acid
- TFE:
-
2,2,2-Trifluoroethanol
- Tol:
-
Toluene
- Trt:
-
Trityl
References
Albericio F, Hammer RP, García-Echevarría C, Molins M-A, Chang J, Munson M, Pons M, Giralt E, Barany G (1991) Cyclization of disulfide-containing peptides in solid-phase synthesis. Int J Pept Protein Res 37:402–413
Alexopoulos K, Fatseas P, Melissari E, Vlahakos D, Roumelioti P, Mavromoustakos T, Mihailescu S, Paredes-Carbajal MC, Mascher D, Matsoukas J (2004) Design and synthesis of novel biologically active thrombin receptor non-peptide mimetics based on the pharmacophoric cluster Phe/Arg/NH2 of the Ser42-Phe-Leu-Leu-Arg46 motif sequence: platelet aggregation and relaxant activities. J Med Chem 47(13):3338–3352
Bacsa B, Bösze S, Kappe CO (2010) Direct solid-phase synthesis of the β-amyloid (1–42) peptide using controlled microwave heating. J Org Chem 75:2103–2106
Barlos K, Gatos D, Kapolos S, Papaphotiu G, Schafer W, Wenqing Y (1989) Esterification of partially protected peptide-fragments with resins—utilization of 2-chlorotritylchloride for synthesis of Leu-15-gastrin-I. Tetrahedron Lett 30:3947–3950
Bernard-Pierrot I, Delbe J, Caruelle D, Barritault D, Courty J, Milhiet PE (2001) The lysine-rich C-terminal tail of heparin affin regulatory peptide is required for mitogenic and tumor formation activities. J Biol Chem 276:12228–12234
Caddick S, Fitzmaurice R (2009) Microwave enhanced synthesis. Tetrahedron 65(17):3325–3355
Chan WC, White PD (2000) Fmoc solid phase peptide synthesis: a practical approach. Oxford University Press Inc., New York
Ducès A, Karaky R, Martel-Renoir D, Mir L, Hamma-Kourbali Y, Biéche I, Opolon P, Delbé J, Courty J, Perricaudet M, Griscelli F (2008) 16-kDa fragment of pleiotrophin acts on endothelial and breast tumor cells and inhibits tumor development. Mol Cancer Ther 7(9):2817–2827
Felix AM, Heimer EP, Wang C-T, Lambros TJ, Swistok J, Roszkowski M, Ahmad M, Confalone D, Scott JW, Parker D, Meienhofer J, Trzeciak A, Gillessen D (1985) Synthesis of thymosin alpha-1 by fragment condensation using tert-butyl side chain protection. Int J Pept Protein Res 26:130–148
Finley EL, Dillon J, Crouch RK, Schey KL (1998) Identification of tryptophan oxidation products in bovine alpha-crystallin. Protein Sci 7(11):2391–2397
Friligou I, Agelis G, Matsoukas J, Tselios T (2008) Efficient microwave-assisted synthesis of myelin epitopes MOG35-55 and MOG97-108 using CLTR-CL resin. J Pept Sci 14(S1):90
Galanis AS, Albericio F, Grøtli M (2008) Enhanced microwave-assisted method for on-bead disulfide bond formation: Synthesis of α-conotoxin MII. Peptide Science 92(1):23–34
Galanis AS, Albericio F, Grøtli M (2009) Solid-phase peptide synthesis in water using microwave-assisted heating. Org Lett 11(20):4488–4491
Góngora-Benítez M, Tulla-Puche J, Paradís-Bas M, Werbitzky O, Giraud M, Albericio F (2010) Optimized FMOC solid-phase synthesis of the Cysteine-rich peptide linaclotide. Biopolymers (Pept Sci). doi:10.1002/bip.21480
Hamma-Kourbali Y, Bernard-Pierrot I, Heroult M, Dalle S, Caruelle D, Milhiet PE, Fernig DG, Delbé J, Courty J (2008) Inhibition of the mitogenic, angiogenic and tumorigenic activities of pleiotrophin by a synthetic peptide corresponding to its C-thrombospondin repeat-I domain. J Cell Physiol 214:250–259
Han Y, Albericio F, Barany G (1997) Occurrence and minimization of cysteine racemization during stepwise solid-phase peptide synthesis. J Org Chem 62:4307–4312
Hayes BL (2002) Microwave synthesis: chemistry at the speed of light. CEM Publishing, Matthews, NC
Heggemann C, Budke C, Schomburg B, Majer Z, Wißbrock M, Koop T, Sewald N (2010) Antifreeze glycopeptide analogues: microwave-enhanced synthesis and functional studies. Amino Acids 38:213–222
Héroult M, Bernard-Pierrot I, Delbé J, Hamma-Kourbali Y, Katsoris P, Barritault D, Papadimitriou E, Plouet J, Courty J (2004) Heparin affin regulatory peptide binds to vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. Oncogene 23:1745–1753
Hulmes JD, Seddon AP, Decker MM, Böhlen P (1993) Comparison of the disulfide bond arrangements of human recombinant and bovine brain heparin binding neurite-promoting factors. Biochem Biophys Res Commun 192:738–746
Inui T, Nakao M, Nishio H, Nishiuchi Y, Kojima S, Muramatsu T, Kimura T (2000) Solution synthesis and biological activity of human pleiotrophin, a novel heparinbinding neurotrophic factor consisting of 136 amino acid residues with five disulfide bonds. J Peptide Res 55:384–397
Isidro-Llobet A, Álvarez M, Albericio F (2009) Amino acid-protecting groups. Chem Rev 109:2455–2504
Kamber B (1973) Die gezielte Synthese offenkettiger asymmetrischer Cystinpeptide mittels thiol-induzierter Fragmentierung von Sulfenylthiocarbonaten. Insulinfragmente mit intakter Disulfibrücke A20-B19. Helv Chim Acta 56:1370–1381
Kamber B, Hartmann A, Eisler K, Riniker B, Rink H, Sieber P, Rittel W (1980) The synthesis of cystine peptides by iodine oxidation of S-trityl-cysteine and S-acetamidomethyl-cysteine peptides. Helv Chim Acta 63:899–915
Kappe CO (2004) Controlled microwave heating in modern organic synthesis. Angew Chem Int Ed 43:6250–6284
Kates SA, Albericio F (2000) Solid phase synthesis: a practical guide. Marcel Dekker, New York
Katsara M, Deraos G, Tselios T, Matsoukas MT, Friligou I, Matsoukas J, Apostolopoulos V (2009) Design and synthesis of a cyclic double mutant peptide (cyclo(87–99)[A91, A96]MBP87–99) induces altered responses in mice after conjugation to mannan: implications in the immunotherapy of multiple sclerosis. J Med Chem 52(1):214–218
Kent SB, Alewood D, Alewood P, Baca M, Jones A, Schnolzer M (1992) Total chemical synthesis of proteins: evolution of solid phase synthetic methods illustrated by the total chemical syntheses of the HIV-1 protease. In: Epton R (ed) Innovation and perspectives in solid phase synthesis. Intercept Ltd. Andover, UK, pp 1–22
Kilpelainen I, Kaksonen M, Kinnunen T, Avikainen H, Fath M, Linhardt RJ, Raulo E, Rauvala H (2000) Heparin-binding growth-associated molecule contains two heparin-binding beta-sheet domains that are homologous to the thrombospondin type I repeat. J Biol Chem 275:13564–13770
Lamthanh H, Roumestand C, Deprun C, Menez A (1993) Side reaction during the deprotection of (S-acetamidomethyl)cysteine in a peptide with a high serine and threonine content. Int J Pept Protein Res 41(1):85–95
Lamthanh H, Virelizier H, Frayssinhes D (1995) Side reaction of S-to-N acetamidomethyl shift during disulfide bond formation by iodine oxidation of S-acetamidomethyl-cysteine in a glutamine-containing peptide. Pept Res 8:316–320
Matsoukas JM, Agelis G, Hondrelis J, Yamdagni R, Wu Q, Ganter R, Moore D, Moore GJ, Smith JR (1993) Synthesis and biological activities of angiotensin II, sarilesin, and sarmesin analogs containing Aze or Pip at position 7. J Med Chem 36(7):904–911
Matsoukas JM, Hondrelis J, Agelis G, Barlos K, Gatos D, Ganter R, Moore D, Moore GJ (1994) Novel synthesis of cyclic amide-linked analogs of angiotensins II and III. J Med Chem 37(18):2958–2969
Matsoukas J, Panagiotopoulos D, Keramida M, Mavromoustakos T, Yamdagni R, Qiao W, Moore G, Saifeddine M, Hollenberg M (1996) Synthesis and contractile activities of cyclic thrombin receptor-derived peptide analogues with a Phe-Leu-Leu-Arg motif: importance of the Phe/Arg relative conformation and the primary amino group for activity. J Med Chem 39:3585–3591
Mikelis C, Koutsioumpa M, Papadimitriou E (2007) Pleiotrophin as a possible new target for angiogenesis-related diseases and cancer. Recent Pat Anticancer Drug Discov 2:175–186
Nagata K, Maruyama K, Nagasawa H, Urushibata I, Isogai A, Ishizaki H, Suzuki A (1992) Bombyxin-II and its disulfide bond isomers: synthesis and activity. Peptides 13:653–662
Ori A, Free P, Courty J, Wilkinson MC, Fernig DG (2009) Identification of heparin-binding sites in proteins by selective labeling. Mol Cell Proteomics 8:2256–2265
Palasek SA, Cox ZJ, Collins JM (2007) Limiting racemization and aspartimide formation in microwave-enhanced Fmoc solid phase peptide synthesis. J Pept Sci 13:143–148
Papadimitriou E, Mikelis C, Lampropoulou E, Koutsioumpa M, Theochari K, Tsirmoula S, Theodoropoulou C, Lamprou M, Sfaelou E, Vourtsis D, Boudouris P (2009) Roles of pleiotrophin in tumor growth and angiogenesis. Eur Cytokine Netw 20(4):180–190
Polykratis A, Delbé J, Courty J, Papadimitriou E, Katsoris P (2004) Identification of heparin affin regulatory peptide domains with potential role on angiogenesis. Int J Biochem Cell Biol 36:1954–1966
Raulo E, Tumova S, Pavlov I, Pekkanen M, Hienola A, Klankki E, Kalkkinen N, Taira T, Kilpelaïnen I, Rauvala H (2005) The two thrombospondin type I repeat domains of the heparin-binding growth-associated molecule bind to heparin/heparan sulfate and regulate neurite extension and plasticity in hippocampal neurons. J Biol Chem 280:41576–41583
Riddles PW, Blakeley RL, Zerner B (1979) Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid): a reexamination. Anal Biochem 94:75–81
Rizzolo F, Sabatino G, Chelli M, Rovero P, Papini AM (2007) A convenient microwave-enhanced solid-phase synthesis of difficult peptide sequences: case study of gramicidin A and CSF114(Glc). Int J Pept Res Ther 13:203–208
Sabatino G, Papini AM (2008) Advances in automatic, manual and microwave-assisted solid-phase peptide synthesis. Curr Opin Drug Discov Devel 11(6):762–770
Seddon AP, Hulmes JD, Decker MM, Kovesdi I, Fairhurst JL, Backer J, Dougher-Vermazen M, Böhlen P (1994) Refolding and characterization of human recombinant heparin-binding neurite-promoting factor. Protein Expr Purif 5:14–21
Sewald N, Jakubke Η-D (2002) Peptides: chemistry and biology. Wiley-VCH Verlag GmbH & Co, KGaA, Germany
Sieber P (1977) Der 2-trimethylsilyläthyl-rest als selektiv abspaltbare carboxy-schutzgruppe. Helv Chim Acta 60:2711–2716
Spyranti Z, Dalkas GA, Spyroulias GA, Mantzourani ED, Mavromoustakos T, Friligou I, Matsoukas JM, Tselios TV (2007) Putative bioactive conformations of amide linked cyclic myelin basic protein peptide analogues associated with experimental autoimmune encephalomyelitis. J Med Chem 50(24):6039–6047
Tam JP, Wu C-R, Liu W, Zhang J-W (1991) Disulfide bond formation in peptides by dimethyl sulfoxide. J Am Chem Soc 113:6657–6662
Vasileiou Z, Barlos KK, Gatos D, Adermann K, Deraison C, Barlos K (2010) Synthesis of the proteinase inhibitor LEKTI domain 6 by the fragment condensation method and regioselective disulfide bond formation. Biopolymers 94(3):339–349
White P, Collins J, Cox Z (2005) Comparative study of conventional and microwave assisted synthesis. In: Poster presentation at the 19th American peptide symposium, San Diego, CA
Yamashiro D, Blake J, Li CH (1976) The use of trifluoroethanol for improved coupling in solid-phase peptide synthesis. Tetrahedron Lett 18:1469–1472
Acknowledgments
This work was supported by the E.U.-European Social Fund (75%) and the Greek Ministry of Development-GSRT (25%) (Grant PENED2003, 036Δ560). Special thanks to Eldrug SA for providing access to CEM Liberty automated microwave peptide synthesizer.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Friligou, I., Papadimitriou, E., Gatos, D. et al. Microwave-assisted solid-phase peptide synthesis of the 60–110 domain of human pleiotrophin on 2-chlorotrityl resin. Amino Acids 40, 1431–1440 (2011). https://doi.org/10.1007/s00726-010-0753-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0753-6