
Abstract
Previously, we found that advanced glycation endproducts (AGEs) directly interact with tumor necrosis factor (TNF)-like weak inducer of apoptosis, a cytokine that controls inflammation, and that this interaction inhibited its action. This finding raised the novel possibility that AGEs alter the function of other cytokines through direct interaction. To investigate this possibility, we performed comprehensive screening for candidates that interacted with AGEs using protein array analysis. The array analysis revealed that high mobility group box-1 (HMGB1) had a markedly high affinity for AGEs. HMGB1 is a representative proinflammatory damage-associated molecular pattern molecule, and is reported to interact with lipopolysaccharide (LPS) directly to exert its inflammatory function. When LPS, HMGB1, and AGEs were mixed, the mobility of HMGB1 had shifted significantly in native PAGE, suggesting that these three molecules formed a triplet complex. The addition of AGEs to the LPS–HMGB1 mixture synergistically potentiated LPS–HMGB1-stimulated TNF-α mRNA expression in macrophage-like RAW264.7 cells. In addition, using receptor knockout clones, the increased proinflammatory response by LPS–HMGB1–AGEs complex was demonstrated to be mediated via Toll-like receptor 4 and receptor for AGEs. Taken together, this study suggested that AGEs carry out their pathophysiological roles by potentiating the LPS–HMGB1-stimulated proinflammatory response through direct interactions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Advanced glycation end products (AGEs) were initially found as the cause of food browning. These molecules are formed in vivo from reducing sugar or its metabolites with the free amino groups of endogenous molecules such as proteins and lipids. Furthermore, the accumulation of AGEs in vivo was suggested to be involved in the pathogenesis of aging-related diseases, hyperglycemia, and some inflammatory diseases such as diabetes and cardiovascular disease [1,2,3,4,5].
As the mechanisms by which AGEs function in the pathogenesis, two possibilities have been proposed. When AGEs are formed on functional proteins, such as laminin, tubulin, or insulin, the function of glycated proteins was mainly reported to be impaired by structural modifications [6,7,8]. In addition, AGEs are believed to elicit inflammatory responses through the stimulation of endogenous receptors, including the receptor for AGEs (RAGE) [9]. Using primary cells from RAGE-knockout (KO) mice, it was revealed that AGE–RAGE interaction activates the NF-κB pathway, proinflammatory cytokine expression, NADPH oxidase activation, and reactive oxygen species production [10, 11]. These findings suggest that AGEs play an essential role in the pathogenesis of several inflammatory diseases such as diabetes.
Previously, we found that AGEs alter cytokine function through direct interaction. Briefly, AGEs directly interact with tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), and this interaction inhibits the function of TWEAK, altering the TNF-α-induced inflammatory response [12]. Moreover, it was found that AGEs can interact with interleukin (IL)-8, a chemotactic cytokine for neutrophils. These findings suggested that AGEs specifically interact with functional proteins and may influence their functions. This suggests an undefined third mechanism in the pathogenesis of AGE-induced inflammatory diseases.
In the present study, to explore potential partner proteins capable of interacting with AGEs, we performed a comprehensive protein array analysis of 487 human target molecules. As a result, high mobility group box-1 (HMGB1) was found to interact with AGEs with high affinity. In addition, the direct interaction among AGEs, HMGB1, and lipopolysaccharide (LPS) synergistically potentiated the proinflammatory responses in macrophage-like RAW264.7 cells compared with their activity alone. The pathophysiological significance of this phenomenon is discussed.
Materials and methods
Screening AGE–protein interactions by protein array
To prepare probes for protein array analysis, bovine serum albumin (BSA) was labeled by biotin using EZ-Link Hydrazide-Biotin (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. A part of this biotin-labeled BSA was incubated with glyceraldehyde to prepare biotin-labeled glyceraldehyde-modified BSA (biotin-labeled AGEs) according to the previously described method [12]. The biotin-labeled AGEs (formed using 100 µγ biotin-labeled BSA) and the biotin-labeled BSA (100 µγ) were used as probes for protein array analysis using Protein Array G2 PAH-G2-2 (RayBiotech, Peachtree Corners, GA, USA). Array analysis was carried out according to the manufacturer’s protocol. Briefly, the biotin-labeled probe protein was incubated with the array on which 487 human target proteins were immobilized. After washing away unbound probe protein, Cy3-Streptavidin was incubated with the array. Subsequently, after washing away unbound Cy3-Streptavidin, Cy3 signals representing the protein interacting with the probe protein were detected and quantified using a laser scanner by Filgen (Nagoya, Japan).
Expression and purification of recombinant HMGB1
In order to construct the N-terminally His-tagged human HMGB1 expression vector for the ExpiSf Baculovirus Expression System (Thermo Fisher Scientific, Waltham, MA, USA), the cDNA encoding HMGB1 was amplified by PCR from the cDNA prepared from human THP-1 cells using forward (5’- CGCCATATGGGCAAAGGAGATCCTAAG) and reverse (5’- CGCGGATCCTTATTCATCATCATC) primers. The amplicon was subcloned into the pCold II vector (Takara Bio, Kusatsu, Japan) at NdeI and BamHI sites to add the His-tag sequence. N-terminally His-tagged human HMGB1 was amplified with this vector using forward (5’- TACGCGGAATTCATGCATCATCATCATCATCATATG) and reverse (5’- AGGCGCAAGCTTTTATTCATCATCATCATCTTCT) primers, and subcloned into the pFastBac 1 vector (Thermo Fisher Scientific, Waltham, MA, USA) at BamHI and EcoRI sites. This vector was used to generate recombinant HMGB1 using the ExpiSf Baculovirus Expression System. Briefly, the vector was introduced into DH10Bac Competent Cells (Thermo Fisher Scientific, Waltham, MA, USA) to generate recombinant Bacmid, and this Bacmid was introduced into ExpiSf9 insect cells to generate high-titer recombinant Baculovirus. The recombinant HMGB1 protein was expressed by infecting Sf9 cells with this virus.
Recombinant His-tagged HMGB1 expressed in the Sf9 cells was purified using Cobalt-bound beads (Takara Bio, Kusatsu, Japan). The endotoxin level was confirmed to be less than 0.01 EU/µg protein using the ToxinSensor Single Test Kit (Genscript, Piscataway, NJ, USA).
Confirmation of interaction among LPS, HMGB1, and AGEs
LPS was purchased from InvivoGen (San Diego, CA, USA). Glyceraldehyde-modified BSA (AGEs) was prepared as previously described [12]. The endotoxin level was confirmed to be less than 0.1 EU/mg protein. C-terminally His-tagged recombinant mouse osteopontin (OPN) was purchased from BioLegend (San Diego, CA, USA).
LPS, recombinant HMGB1 or OPN, and AGEs were mixed and incubated at 37°C for 1 h, and the mixture was separated by native PAGE. The band pattern of HMGB1 or OPN was analyzed by Western blotting using horseradish peroxidase-linked anti-His-tag antibody (Medical & Biological Laboratories, Nagoya, Japan) according to the method previously described [12].
Cell culture, and analyzing the response to LPS, HMGB1, and/or AGEs
RAW264.7 (wild type, WT) cells and previously generated receptor KO clones (TLR4-KO and RAGE-KO) [13] were maintained in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal calf serum (Gibco, Carlsbad, CA, USA), 100 U/mL of penicillin, and 100 µg/mL of streptomycin (Nakalai tesque, Kyoto, Japan) at 37°C in humidified air containing 5% CO2.
The WT cells were stimulated by the mixture of LPS, HMGB1, and AGEs. Then, total RNA was isolated from the cells using the total RNA isolation kit. The concentration and purity of the extracted total RNA were assessed using the 260/280 nm absorbance ratio. Using these RNA samples, real-time RT-PCR was performed as previously described [12]. Similarly, the culture medium of the cells was harvested and analyzed by multiplex ELISA using BD Cytometric Bead Array kit (BD Biosciences, San Jose, CA, USA) with the aid of a NovoCyte Flow Cytometer (ACEA Biosciences, San Diego, CA, USA). In brief, the culture medium was incubated with the mixture of capture beads coated with the antibody for TNF-a, monocyte chemotactic protein (MCP)-1, IL-1β, IL-6, KC, IL-10, or interferon-γ. Each capture bead had a unique fluorescence intensity. The proteins captured by the beads were subsequently incubated with the mixture of the detection antibodies specific for each protein. The detection antibodies were conjugated with phycoerythrin (PE). Using a flow cytometer, the beads were then separated based on the unique fluorescence of the beads, and the protein amount was estimated based on the PE fluorescence intensity. Obtained data were analyzed to calculate protein concentration using FCAP Array Software (version 3.0, BD Biosciences, San Jose, CA, USA). The culture medium of the WT cells and the KO cells stimulated by the mixture of LPS, HMGB1, and AGEs were analyzed by conventional ELISA using LEGEND MAX Mouse TNF-α ELISA Kit (BioLegend, San Diego, CA, USA) according to the manufacturer’s protocol.
Statistical analyses
Statistical analyses were performed hypothesizing normality and homoscedasticity of the data, using the Student’s t-test for 2 groups or the Tukey-Kramer test for multiple groups. Analyses were performed using R (version 3.6.1, The R Foundation for Statistical Computing, Vienna, Austria). P-values < 0.05, 0.01 or 0.001 were considered significant. All data are presented as the mean ± standard error (SE).
Results
HMGB1 interacted with LPS and/or AGEs to form complexes
To explore novel proteins that interact with AGEs, we performed comprehensive screening using protein array analyses. The array was incubated with biotin-labeled AGEs or BSA probes, which interacted with target proteins immobilized on the array and were detected by Cy3-Streptavidin. Cy3 signals were quantified by a laser scanner, and signal values were calculated by subtracting the fluorescence intensity on the BSA-treated array from that on the AGEs-treated array. Among the 487 target proteins on the array, HMGB1 produced the highest signal value, suggesting a marked interaction between AGEs and HMGB1 (Table 1).
This result was confirmed by the separation experiment of the HMGB1 and AGE mixture using native PAGE. As HMGB1 has also been reported to interact with LPS [14, 15], the possibility of formation of LPS–HMGB1 complexes or LPS–HMGB1–AGEs complexes was analyzed on native PAGE. As shown in Fig. 1a, the mobility and signal intensity of HMGB1 in the mixture of LPS and HMGB1 were largely changed compared with those of HMGB1 alone, suggesting the interaction between LPS and HMGB1. Furthermore, the change to a smear band pattern was noted in the mixture of HMGB1 and AGEs, confirming their interaction. In addition, when LPS, HMGB1, and AGEs were mixed, a similar band shift was observed, suggesting the formation of complexes comprising the three molecules.
In the array result, osteopontin (OPN) was found as one of the proteins yielded a negative signal value, suggesting that this protein did not interact with AGEs. When HMGB1 was replaced with OPN, the band patterns of OPN were not changed in any mixtures (Fig. 1b), suggesting that OPN did not interact with LPS or AGEs.

Interaction among LPS, HMGB1, and AGEs. a Mixtures of 500 ng of HMGB1 with and without and 100 ng of LPS and/or 2.5 µg of AGEs were incubated for 1 h, and separated by native PAGE. The band pattern of HMGB1 was detected and compared by Western blotting. b OPN (100 ng) was analyzed as a negative control for HMGB1 by the same procedure as a
LPS, HMGB1, and AGEs synergistically stimulated the expression of proinflammatory cytokines in RAW264.7 cells
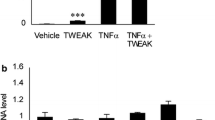
To clarify the physiological implications for the interaction among LPS, HMGB1, and AGEs, we next examined the changes in proinflammatory activity based upon the complex formation. When macrophage-like RAW264.7 cells were stimulated with 10 pg/mL of LPS (Fig. 2a), 100 ng/mL of HMGB1 (Fig. 2b), or 2 µg/mL of AGEs (Fig. 2c) alone, the expression of proinflammatory TNF-α was hardly affected. Subsequently, the cells were stimulated by the combination of two molecules at the same concentration. As a result, the mixture of LPS and HMGB1 or LPS and AGEs significantly induced TNF-α expression (Fig. 2d, f), whereas HMGB1 and AGEs had no stimulatory effects (Fig. 2e). In addition, TNF-α mRNA expression stimulated by the mixture of LPS, HMGB1, and AGEs was far higher than that by the mixture of LPS and HMGB1, being 4.7-times and 2.5-times higher than that by vehicle, respectively (Fig. 2g).

The mixture of LPS, HMGB1, and AGE stimulated TNF-α gene expression in RAW264.7 cells. RAW264.7 cells were stimulated with vehicle, 10 pg/mL of LPS (a), 100 ng/mL HMGB1 (b), or 2 µg/mL of AGEs (c) for 1 h, and TNF-α gene expression was then measured by real-time RT-PCR. The TNF-α mRNA levels were normalized against the β-actin mRNA levels. (d–f) Similar to (a–c), the cells were stimulated with vehicle, LPS and HMGB1, HMGB1 and AGEs, or LPS and AGEs, and TNFα mRNA levels were measured. (g) Similar to (a–f), the cells were stimulated with vehicle, LPS and HMGB1, or LPS, HMGB1, and AGEs, and TNF-α mRNA levels were measured. ***, P < 0.001; *, P < 0.05 versus vehicle by Student’s t-test (A–F) (n = 03). ***, P < 0.001 by Tukey-Kramer test (G) (n = 03)
To support these synergistic effects at the protein level, the change in the levels of TNF-α and other proinflammatory cytokines in culture medium was measured by multiplex ELISA. As shown in Fig. 3a, TNF-α concentrations in the culture medium of vehicle-, LPS-, LPS and HMGB1-, or LPS, HMGB1 and AGE-stimulated cells increased sequentially, analogous to the results shown in Fig. 2. In addition, a similar change was observed in MCP-1 (Fig. 3b). Other cytokines, including IL-1β, IL-6, KC (the murine IL-8 homolog), IL-10, and interferon-γ, were hardly detectable in the culture medium of all stimulated cells (data not shown).

The mixture of LPS, HMGB1, and AGE stimulated TNF-α and MCP-1 secretion in RAW264.7 cells. RAW264.7 cells were stimulated with vehicle, 10 pg/mL of LPS, LPS and 100 ng/mL HMGB1, or LPS, HMGB1, and 2 µg/mL of AGEs for 12 h, and then the TNF-α a and MCP-1 b protein concentrations in culture medium were measured by multiplex ELISA. ***, P < 0.001; *, P < 0.05 by Tukey-Kramer test (n = 03)
TLR4 and RAGE function in the increased proinflammatory response stimulated by LPS, HMGB1, and AGEs
We next focused on revealing the receptors responsible for the increased proinflammatory response by LPS, HMGB1, and AGE. For this purpose, two receptor KO clones, Toll-like receptor 4 (TLR4)-KO clone and RAGE-KO clone in RAW264.7 cells [13], were used. The synergistic effects by the combination of LPS, HMGB1, and AGEs on TNF-α production were almost abolished in the TLR4-KO clone compared with wild type cells (Fig. 4a, b). In the RAGE-KO clone, a slight decrease in TNF-α production was noted (Fig. 4c). These results suggested that the combination of LPS, HMGB1, and AGE induced cellular responses via stimulation of TLR4 and RAGE.

The mixture of LPS, HMGB1, and AGE stimulated TNF-α secretion in RAW264.7 cells or receptor-KO clones. The WT cells (a), TLR4-KO clone (b), and RAGE-KO clone (c) were stimulated with vehicle or a mixture of 10 pg/mL of LPS, 100 ng/mL HMGB1, and 2 µg/mL of AGEs for 12 h, and the TNF-α protein concentrations in culture medium were measured by ELISA. For each cell, the TNF-α protein levels are shown as ratios normalized against the TNF-α protein concentration in the culture medium of vehicle-stimulated cells. ***, P < 0.001; *, P < 0.01 versus vehicle by Student’s t-test (n = 03)
Discussion
We previously found that AGEs altered the function of TWEAK through their direct interaction [12]. This suggested that AGEs have a unique property to change the function of cytokines by interacting with them. To investigate this, in the present study, protein array analyses were employed to comprehensively screen for protein candidates capable of interacting with AGEs.
Among the 487 target proteins, HMGB1 was found to be capable of interacting with AGEs with the highest affinity. HMGB1 is a nucleoprotein well-known as a damage-associated molecular pattern (DAMP) molecule. DAMP molecules are endogenous proteins normally located inside of cells. When cells were disrupted by injury, infection, or inflammatory reactions, DAMP molecules translocate to the outside and act as proinflammatory factors [16]. At present, it remains controversial whether DAMP molecules exhibit their functions individually or by interacting with other factors. In the case of HMGB1, it was reported that its coexistence and interaction with LPS were necessary for its proinflammatory activity [14, 15]. As AGEs are also known to function as proinflammatory factors, they may interact with LPS, form complexes, similar to HMGB1, and then stimulate inflammatory responses synergistically.
To evaluate this possibility, we examined the interactions among LPS, HMGB1, and AGEs using native PAGE. The mobility of the band corresponding to HMGB1 was changed when HMGB1 was premixed with LPS or AGEs (Fig. 1a). This suggested that the combination of LPS, HMGB1, and AGEs is capable of forming complexes. In addition, a difference in the band intensity of HMGB1 was observed when HMGB1 was mixed with LPS or AGEs, suggesting diverse sizes and charges. Although the details of this phenomenon are unknown, this may be caused by the diverse structures of LPS or AGEs.
As OPN hardly interacted with AGEs in the array analysis, its interaction with LPS or AGEs was evaluated as a negative control for HMGB1. As shown in Fig. 1b, OPN was unable to interact with LPS or AGEs, suggesting that LPS or AGEs specifically interacted with HMGB1.
In this study, formation of these complexes was found to alter the proinflammatory activity of LPS, HMGB1, and AGEs. The mixture of LPS and HMGB1 stimulated TNF-α expression (Fig. 2d), although the same amounts of these molecules alone had little or no effect, respectively (Fig. 2a, b). Similarly, the mixture of LPS and AGEs demonstrated synergic effects on TNF-α expression (Fig. 2f), although the mixture of HMGB1 and AGEs had no effect (Fig. 2e). Furthermore, the combination of AGEs, LPS, and HMGB1 synergistically increased TNF-α expression (Fig. 2g). These results were confirmed by measurement of the TNF-α production at the protein level (Fig. 3).
Using receptor-KO clones, the complex-stimulated TNF-α expression was suggested to be induced through TLR4 and RAGE. In brief, TNF-α expression stimulated by the complexes comprising AGEs, LPS, and HMGB1 was almost completely or moderately suppressed in TLR4- or RAGE-KO cells, respectively (Fig. 4). TLR4 is a well-known LPS receptor, and was reported to be involved in HMGB1 recognition [16]. In addition, we previously found that TLR4 functioned in AGE recognition [13]. Moreover, RAGE is believed to be responsible for both AGEs and HMGB1 recognition [16]. This suggested that the complexes comprising LPS, HMGB1, and AGEs stimulate their corresponding receptors.
Synergic inflammatory action by LPS, HMGB1, and AGEs may be correlated with the exacerbation of inflammatory response in diabetic patients. It was previously reported that the inflammatory response in RAW264.7 cells was synergistically activated by the combination of histone, HMGB1, and DNA, similar to our study [17]. These three molecules have been defined as DAMP molecules, suggesting their pathophysiological roles in DAMP-related inflammatory diseases. Similarly, we found synergic proinflammatory effects by HMGB1, pathogen-derived LPS, and endogenously generated AGEs in this study. Synergic activation of the inflammatory response by DAMP proteins and pathogen-derived LPS plays a defensive role against infectious diseases. On the other hand, in diabetic patients with excessive AGE accumulation, the formation of LPS–HMGB1–AGE complexes likely causes prolonged inflammatory responses and aggravates the medical condition. To clarify this hypothesis, further investigations using an in vivo inflammatory model are in progress.
In addition, we found several candidate molecules interacted with AGEs using protein array (Table 1). There are possibilities that the functions of these candidates are altered similarly to HMGB1 by the interaction with AGEs. To examine these possibilities, detailed investigations are ongoing.
Data Availability
The data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Aso Y, Inukai T, Tayama K, Takemura Y (2000) Serum concentrations of advanced glycation endproducts are associated with the development of atherosclerosis as well as diabetic microangiopathy in patients with type 2 diabetes. Acta Diabetol 37:87–92
Kanauchi M, Tsujimoto N, Hashimoto T (2001) Advanced glycation end products in nondiabetic patients with coronary artery disease. Diabetes Care 24:1620–1623
Li S-Y, Du M, Dolence EK et al (2005) Aging induces cardiac diastolic dysfunction, oxidative stress, accumulation of advanced glycation endproducts and protein modification. Aging Cell 4:57–64. https://doi.org/10.1111/j.1474-9728.2005.00146.x
Srikanth V, Maczurek A, Phan T et al (2011) Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging 32:763–777. https://doi.org/10.1016/j.neurobiolaging.2009.04.016
Reynaert NL, Gopal P, Rutten EPA et al (2016) Advanced glycation end products and their receptor in age-related, non-communicable chronic inflammatory diseases; Overview of clinical evidence and potential contributions to disease. The International Journal of Biochemistry Cell Biology 81:403–418. https://doi.org/10.1016/j.biocel.2016.06.016
Kaji Y, Usui T, Oshika T et al (2000) Advanced glycation end products in diabetic corneas. Investig Ophthalmol Vis Sci 41:362–368
Williams SK, Howarth NL, Devenny JJ, Bitensky MW (1982) Structural and functional consequences of increased tubulin glycosylation in diabetes mellitus. Proc Natl Acad Sci USA 79:6546–6550. https://doi.org/10.1073/pnas.79.21.6546
Hunter SJ, Boyd AC, O’Harte FP et al (2003) Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes 52:492–498
Ott C, Jacobs K, Haucke E et al (2014) Role of advanced glycation end products in cellular signaling. Redox Biology 2:411–429. https://doi.org/10.1016/j.redox.2013.12.016
Liliensiek B, Weigand MA, Bierhaus A et al (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113:1641–1650. https://doi.org/10.1172/JCI200418704
Tadié J-M, Bae H-B, Banerjee S et al (2012) Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am J Physiol Cell Physiol 302:C249–C256. https://doi.org/10.1152/ajpcell.00302.2011
Watanabe M, Toyomura T, Wake H et al (2017) Advanced glycation end products attenuate the function of tumor necrosis factor-like weak inducer of apoptosis to regulate the inflammatory response. Mol Cell Biochem 434:153–162. https://doi.org/10.1007/s11010-017-3045-6
Watanabe M, Toyomura T, Wake H et al (2019) Differential contribution of possible pattern-recognition receptors to advanced glycation end product-induced cellular responses in macrophage-like RAW264.7 cells. Biotechnol Appl Biochem. https://doi.org/10.1002/bab.1843
Hreggvidsdóttir HS, Östberg T, Wähämaa H et al (2009) The alarmin HMGB1 acts in synergy with endogenous and exogenous danger signals to promote inflammation. J Leukoc Biol 86:655–662. https://doi.org/10.1189/jlb.0908548
Hreggvidsdóttir HS, Lundberg AM, Aveberger A-C et al (2012) High mobility group box protein 1 (HMGB1)-partner molecule complexes enhance cytokine production by signaling through the partner molecule receptor. Mol Med 18:224–230. https://doi.org/10.2119/molmed.2011.00327
Roh JS, Sohn DH (2018) Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw 18:e27. https://doi.org/10.4110/in.2018.18.e27
Chen R, Fu S, Fan X-G et al (2015) Nuclear DAMP Complex-mediated RAGE-Dependent Macrophage Cell Death. Biochem Biophys Res Commun 458:650–655. https://doi.org/10.1016/j.bbrc.2015.01.159
Acknowledgements
This study was supported by JSPS KAKENHI Grant Numbers 18K06807 and 18K14969, and the Wesco Scientific Promotion Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest associated with this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Watanabe, M., Toyomura, T., Tomiyama, M. et al. Advanced glycation end products (AGEs) synergistically potentiated the proinflammatory action of lipopolysaccharide (LPS) and high mobility group box-1 (HMGB1) through their direct interactions. Mol Biol Rep 47, 7153–7159 (2020). https://doi.org/10.1007/s11033-020-05783-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05783-y