
Abstract
Taurine effectively prevents ischemia-induced apoptosis in the cardiomyocytes and hypothalamic nuclei. The present study explores the influence of taurine on mitochondrial damage, oxidative stress and apoptosis in experimental liver fibrosis. Male albino Wistar rats were divided into six groups and maintained for a period of 60 days as follows: Group I, control; Group II, ethanol treatment [6 g/(kg/day)]; Group III, fibrosis induced by ethanol and iron (0.5% w/w); Group IV, ethanol + iron + taurine (2% w/v); Group V, ethanol + taurine treatment and Group VI, control + taurine treatment. Hepatocytes isolated from ethanol plus iron-treated rats showed decreased cell viability and redox ratio, increased reactive oxygen species formation, lipid peroxidation, DNA fragmentation, and formation of apoptotic bodies. Liver mitochondria showed increased susceptibility to swell, diminished activities of mitochondrial respiratory chain complexes and antioxidants. Taurine administration to fibrotic rats restored mitochondrial function, reduced reactive oxygen species formation, prevented DNA damage, and apoptosis. Thus taurine might contribute to the amelioration of the disease process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic ethanol administration in experimental animals significantly increases the percentage of apoptotic hepatocytes (Yacoub et al. 1995). A link between ethanol metabolism, cytochrome P450 isoenzyme (CYP2E1) induction, enhanced reactive oxygen species (ROS) formation, lipid peroxidation, and cytotoxicity of apoptotic nature has been actively studied and documented (Lieber 1997). Impairment of mitochondrial function is a prelude to the apoptotic event. Mitochondrial abnormalities induced by ethanol are disruption of electron transport chain, oxidative phosphorylation, and ATP generation, all of which are believed to result from increased ROS production (Green and Reed 1998). García-Ruiz et al. (1994) observed loss of functional integrity of mitochondria as a cause of increased ROS production which contributes to apoptotic death. Reduced glutathione (GSH) is the most abundant non-protein antioxidant in the cells which plays a pivotal role in the defense against oxidative stress-induced cell injury and mitochondrial damage (Dickinson and Forman 2002). The concentrations of cellular and mitochondrial GSH are reported to be low in ethanol-fed animals (Fernandez-Checa and Kaplowitz 2005).
Taurine (2-aminoethane sulfonic acid) is a conditionally essential, non-protein amino acid present in blood and tissues of mammals. It plays many important roles in the body-like bile acid conjugation, detoxification, membrane stabilization, osmoregulation, calcium homeostasis, and neurotransmission (Huxtable 1992). Many studies suggest that taurine supplementation can lower oxidative stress (Balkan et al. 2001; Schaffer et al. 2003). The liver-protective role of taurine against toxic insult has been shown (Hwang and Wang 2001; Tabassum et al. 2006). In our earlier study, we observed that taurine circumvents oxidative stress and liver pathology in experimental alcoholic liver disease (Pushpakiran et al. 2004, 2005). Taurine has also been reported to have anti-apoptotic properties (Wu et al. 1999; Takatani et al. 2004).
The spectrum of alcoholic liver disease ranges from minimal injury to hepatitis, fibrosis, and cirrhosis. However, studies have observed that rats fed alcohol alone do not develop alcoholic liver disease beyond the stage of fatty liver, the addition of iron exacerbates the oxidative stress induced by alcohol, and rats treated with a combination of iron and alcohol develop liver damage faster than rats given alcohol alone (Caro and Cederbaum 2001). Thus the combination of iron and alcohol as a model of liver fibrosis was chosen. The study investigates whether taurine could ameliorate mitochondrial dysfunction, DNA damage, and apoptosis in liver of fibrotic rats.
Materials and methods
Chemicals
Iron carbonyl, rotenone, coenzyme Q1 and Q2, antimycin A, dodecyl-β-d-maltoside cytochrome c, propidium iodide, RNAase A, and ethidium bromide were purchased from Sigma Aldrich Pvt. Ltd., St. Louis, MO, USA. Taurine was obtained from Sisco Research Laboratories Pvt. Ltd., Mumbai, India. The solvents and other chemicals used were of analytical grade and purchased from Himedia Laboratories Pvt. Ltd., Mumbai, India.
Treatment of animals
Male albino rats of Wistar strain weighing 170–190 g were purchased from the Central Animal House, Rajah Muthiah Medical College, Annamalai University. The animals were housed in polypropylene cages and maintained under controlled conditions of 12 h light/12 h dark cycle and 50% humidity at 25–30°C. The animals were fed pellet diet (Kamadhenu Agencies, Bangalore, India) and water ad libitum. The study was approved by Institutional Animal Ethics Committee (IAEC), Rajah Muthiah Medical College and Hospital, Annamalai University, and the animals were maintained in accordance with the Guide for the Care and Use of Laboratory Animals. After a period of 1 week, the animals were divided into 6 groups of 12 rats each.
-
Group I received glucose (5 g/kg) isocaloric to ethanol from 40% glucose stock solution.
-
Group II received ethanol at a dosage of 3 g/kg from 30% stock solution twice daily [6 g/(kg/day)].
-
Group III received ethanol and iron carbonyl (0.5% w/w).
-
Group IV received ethanol, iron, and taurine (2% w/v).
-
Group V received ethanol and taurine.
-
Group VI received isocaloric glucose and taurine.
Ethanol and glucose were administered by oral gavage. Iron carbonyl was added to the powdered diet at the level of 0.5% w/w (Bacon et al. 1983). Taurine was administered in drinking water (2% w/v) (Anitha Nandhini et al. 2002). Food and water were fed ad libitum to all groups.
At the end of the experimental period (60 days), six rats from each group were fasted overnight, administered ketamine hydrochloride (30 mg/kg bw) and killed. The animals were cut opened, liver was dissected out and washed in ice-cold saline, blotted, weighed, and mitochondria were isolated for further studies.
Preparation of mitochondria, and measurement of electron transport chain activity and mitochondrial swelling
Mitochondria were isolated by differential centrifugation (Uysal et al. 1989). The protein content was measured by the method of Lowry et al. (1951) and purity was checked by assaying succinate dehydrogenase activity (Slater and Bonner 1952). The electron transport chain activity was determined by measuring the mitochondrial activities of complexes I, II, III, and IV. Mitochondrial samples were subjected to three cycles of fast freeze–thaw in hypotonic buffer (20 mM potassium buffer, pH 7.2) prior to the assay. The activities of complexes I, II, and III were assayed as described by Brich-Machin et al. (1993) with a few modifications. The assay mixture for complex I contained 25 mM potassium phosphate buffer, pH 7.2, 5 mM MgCl2, 2 mM potassium cyanide (KCN), 2.5 mg/ml bovine serum albumin, 0.13 mM NADH, 65 μM coenzyme Q1, and 2 μg/ml antimycin A. Mitochondrial sample was added to initiate the reaction and the oxidation was monitored at 340 nm (ε 6.81 mM−1 cm−1) for 1 min. For complex II (succinate dehydrogenase) assay, mitochondrial sample was preincubated in a medium containing 25 mM potassium phosphate buffer, pH 7.2, 5 mM MgCl2, and 20 mM succinate for 10 min at 30°C. Antimycin A (2 μg/ml), rotenone (2 μg/ml), KCN (2 mM), and 2, 6-dichlorophenol indophenol (50 mM) were added and further incubated for 1 min. The reaction was initiated with coenzyme Q1 (65 μM) and the enzyme-catalyzed reduction of 2, 6-dichlorophenol indophenol was recorded for 3 min at 600 nm (ε 19.1 mM−1 cm−1). The activity of complex III (coenzyme Q cytochrome c reductase) was determined by measuring the rate of reduction of cytochrome c (III) by the reduced form of coenzyme Q2 (ubiquinol) at 550 nm (ε 20 mM−1 cm−1). Complex IV (cytochrome c oxidase) assay was carried out by the method of Wharton and Tzagoloff (1967). The measurement is based on the decrease in absorbance at 550 nm by the oxidation of ferrocytochrome by cytochrome c oxidase.
For swelling studies, the medium contained mitochondrial pellet (0.5 mg protein ml−1), 125 mM sucrose, 10 mM HEPES buffer, pH 7.2, 2.5 mM succinate, and 4.0 μM rotenone in a final volume of 1.0 ml. Mitochondrial swelling was evaluated by monitoring the change in absorbance at 520 nm in a spectrophotometer for 5 min. Decreasing values of absorbance indicate mitochondrial swelling (Garlid and Beavis 1985).
Antioxidant enzymes and reduced glutathione status
The activity of superoxide dismutase (SOD, EC.1.15.1.1), catalase (CAT, EC.1.11.1.6), and glutathione peroxidase (GPx, (EC.1.15.1.9) were measured in mitochondria by methods described in detail elsewhere (Anitha Nandhini et al. 2002). Glutathione reductase (GR) was assayed by the method of Horn and Burn (1978). The GSH content in mitochondria was determined according to the method of Tietze (1969).
Isolation of hepatocytes
Hepatocytes were isolated from a set of six rats in each group. Rats were fasted for 24 h and anesthetized with ketamine hydrochloride (30 mg/kg bw, ip), and the hepatocytes were isolated by the collagenase perfusion technique (Seglen 1976) and used for the following assays.
Cell viability and MTT assay
Cell viability was measured by the ability of the living cells to exclude trypan blue vital dye. Cells were washed twice with phosphate-buffered saline (PBS) and trypan blue in PBS was added at a final concentration of 0.2%. Living cells that did not take up the stain were counted in a hemocytometer and expressed as the percentage of the total count. Mitochondrial function was assayed by the following conversion of MTT 3-(4,5-dimethylthiazol-2-yl-2,5-diphenyl-tetrazolium bromide) by mitochondrial dehydrogenases to purple formazan crystals. Factors that inhibit dehydrogenase activity will affect the associated color formation. Hepatocytes were incubated in the dark at 37°C with 50 μl MTT (1 mg/ml) for 4 h. After incubation, the medium was removed, the formazan crystals were dissolved in 200 μl of dimethylsulfoxide (DMSO), and the absorbance was measured at 570 nm. The percentage reaction was calculated assuming 100% reaction for hepatocytes from control rats.
Reactive oxygen species formation, lipid peroxidation and redox status
Intracellular superoxide formation was determined according to the method of Eligini et al. (2005). An aliquot of hepatocyte (0.1 ml) was incubated with 0.5 ml of nitroblue tetrazolium (NBT) (final concentration 1 mg ml−1) for 30 min. The cells were then treated with 0.6 ml of a solution containing 90% DMSO, 0.01 N NaOH, and 0.1% sodium dodecyl sulfate (SDS). Absorbance was measured at 630 nm. NBT reduction correlates with the amount of superoxide (O2−) produced by the cells. Cellular lipid peroxidation was determined by assaying the formation of thiobarbituric acid substance (TBARS) by spectrophotometry (Niehaus and Samuelsson 1968). Briefly, a 0.5 ml aliquot of the cell suspension was added to 2.0 ml a reagent containing trichloroacetic acid:thiobarbituric acid:hydrochloric acid reagent (TCA:TBA:HCl in 1:1:1 ratio). The reaction mixture was heated in a boiling water bath for color development, cooled to room temperature, and centrifuged to remove insoluble materials. The absorbance of clear supernatant was measured at 535 nm. Lipid hydroperoxide (LHP) was determined according to the method of Jiang et al. (1992). An aliquot of hepatocyte (0.1 ml) was incubated with 0.9 ml of FOX reagent for 30 min at room temperature and the absorbance was measured at 560 nm. Reduced and oxidized forms of glutathione (GSH and GSSG) in hepatocytes were estimated by the method of Tietze (1969) and expressed as a ratio (GSH/GSSH). Protein content of the cells was measured by the method of Lowry et al. (1951).
Propidium iodide (PI) staining
Hepatocytes were washed in PBS thrice and incubated for 10 min with 50 μl of PI (5 mg/ml), mounted in slides, and examined by fluorescence microscopy (Olympus BX51).
DNA fragmentation assay
The DNA fragmentation pattern was analyzed by agarose gel electrophoresis. Hepatocytes were collected and centrifuged at 1,000×g for 10 min. The cell pellets were suspended in 1 ml of lysis buffer consisting of 50 mM Tris–HCl, pH 8.0, 10 mM NaCl, 10 mM EDTA, 100 μg/ml proteinase K, and 0.5% SDS and incubated for 1 h at 50°C. 10 μl of 100 μg/ml ribonucleaseA (RNaseA) was added to the mixture and incubated for an additional 1 h at 50°C. DNA samples were treated with 1 ml phenol followed by extraction with chloroform/isoamylalcohol. The aqueous phase was treated with 25–50 μl of 3 M sodium acetate (pH 5.2) and one volume of ethanol, shaken gently, and left at −20°C, overnight. The precipitate was collected by centrifugation at 12,000×g for 20 min. The pellets were rinsed with 1 ml of 70% ethanol and spun for 10 min. The supernatant was discarded and the pellet was air dried at room temperature and later dissolved in 0.5–1.0 ml of double distilled water. DNA fragmentation was assayed by electrophoresing in 1.0% agarose/ethidium bromide gel for 90 min at 90 V and photographed with UV transilluminator.
Statistical analysis
Results are presented as mean ± SD. Data were analyzed by one-way analysis of variance (ANOVA) followed by Duncan’s Multiple Range Test (DMRT) using the statistical software package (SPSS for Windows, V. 13.0, Chicago, IL, USA).
Results
The contents of TBARS, LHP, GSH, GSSG, and redox ratio in rat hepatocytes are given in Table 1. An increase in TBARS and LHP was found in ethanol and ethanol plus iron-treated animals. The increase was more pronounced in hepatocytes isolated from ethanol plus iron-treated rat hepatocytes. Supplementation with taurine lowered the levels of TBARS and LHP. The redox ratio was significantly lower in ethanol and ethanol plus iron-treated rats as compared to control hepatocytes. Treatment with taurine restored the ratio to near control values.
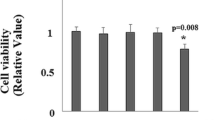
Viability of cells from each group is expressed as percentage of total cells. There was a decrease (47%) in cell viability in ethanol-treated rats and 36% in ethanol plus iron-treated rats (Fig. 1). Taurine treatment prevented cell death induced by ethanol and ethanol plus iron as indicated by the increased cell viability of about 74 and 66%, respectively. Figure 2 illustrates the percentage of MTT metabolism in hepatocytes of animals from various groups. Hepatocytes isolated from ethanol-treated rat showed decreased MTT metabolism (45%) while ethanol plus iron-treated rats showed a further decrease in MTT metabolism (36%). In the presence of taurine, MTT metabolism was increased to 78 and 67% in ethanol and ethanol plus iron treated rats, respectively.

Viability of hepatocytes expressed as percentage (%) of control. Values are means ± SD (n = 6 in each group). Analysis of variance followed by DMRT, P < 0.05. *Significant as compared to control, ¶significant as compared to ethanol, †significant as compared to ethanol + iron. CON control, EtOH ethanol, EtOH + Fe ethanol + iron, EtOH + Fe + TAU ethanol + iron + taurine, EtOH + TAU ethanol + taurine, CON + TAU control + taurine

Effect of taurine on MTT metabolism of experimental groups. Values are means ± SD (n = 6 in each group). Analysis of variance followed by DMRT; P < 0.05. *Significant as compared to control, ¶significant as compared to ethanol, †significant as compared to ethanol + iron. CON control, EtOH ethanol, EtOH + Fe ethanol + iron, EtOH + Fe + TAU ethanol + iron + taurine, EtOH + TAU ethanol + taurine, CON + TAU control + taurine
Intracellular superoxide levels in the hepatocytes obtained from ethanol and ethanol plus iron treated groups compared to those from control animals were significantly higher (Control 13.39 ± 1.02 μmol ml−1; ethanol 23.94 ± 1.82 μmol ml−1; ethanol + iron 32.29 ± 2.46 μmol ml−1). Taurine treatment significantly decreased the level of superoxide by its antioxidant property (ethanol + iron + taurine 20.78 ± 1.58 μmol ml−1; ethanol + taurine 15.05 ± 1.14 μmol ml−1).
Table 2 represents the activities of respiratory chain complexes in liver mitochondria from experimental animals. Statistically significant reduction in the activities of respiratory chain complexes was observed in ethanol, ethanol plus iron-treated rats. Administration of taurine significantly (P < 0.05) increases the values to near-normal.
Table 3 gives the activities of the antioxidant enzymes and glutathione levels in various experimental groups. The enzymes SOD, CAT, GPx, and GR in ethanol plus iron-treated groups showed decreased activity by 35, 49, 40, and 41%, respectively, and GSH by 32% compared to control rats. Taurine administration significantly increased the activities of antioxidant enzymes and GSH levels.
The ability of mitochondria to swell under experimental conditions is a technique that is easy to perform, and is considered to be a sensitive and reliable technique to study the prooxidant-induced permeability of mitochondria. Figure 3 shows the susceptibility of mitochondria to swell. Mitochondria from ethanol group and ethanol plus iron-treated group showed increased swelling as compared to those from control group. The swelling was significantly blunted in TAU-treated group indicating that TAU is able to protect the mitochondrial membrane.

Mitochondrial swelling in ethanol, ethanol plus iron and the effect of taurine. Values are means ± SD (n = 6 in each group). Analysis of variance followed by DMRT, P < 0.05. *Significant as compared to control, ¶significant as compared to ethanol, †significant as compared to ethanol + iron. CON control; EtOH ethanol; EtOH + Fe ethanol + iron; EtOH + Fe +TAU ethanol + iron + taurine; EtOH + TAU ethanol + taurine; CON + TAU control + taurine
Figure 4a–f shows hepatocyte morphology after PI staining. Cells from control animals exhibited healthy nuclei (Fig. 4a, f), while hepatocytes isolated from ethanol, ethanol plus iron treated groups showed highly fluorescent, condensed, and fragmented chromatin suggesting apoptosis mode of cell death (Fig. 4b, c). Treatment with taurine showed regular morphology with round contours and reduced uptake of the dye (Fig. 4d, e).

Propidium iodide staining of isolated rat hepatocytes in EtOH, EtOH + Fe-treated liver cells in the absence and presence of taurine. Apoptotic cells with fragmented nuclei (arrows) are seen in EtOH, EtOH + Fe-treated cells (b, c). Hepatocytes isolated from ethanol, ethanol plus iron-treated rats coadministered with taurine treated-rats (d, e) show reduced apoptotic bodies while hepatocytes from control + taurine (f) show similar morphology as that with control (a). Magnification ×40
The extent of DNA damage in rat hepatocytes was determined by DNA fragmentation assay. The results are shown in Fig. 5. An increased DNA damage and streak formation were observed in ethanol plus iron-treated rats (lane 3) as compared to normal animals (lane 2). Ethanol-treated rats showed decreased DNA damage (lane 4) when compared to ethanol plus iron-treated rats (lane 3). No DNA damage was observed in control (lane 2) and taurine-treated animals (lanes 5–7).

DNA fragmentation pattern in liver of control and experimental animals. Lane 1 molecular weight marker, lane 2 CON, lane 3 EtOH + Fe, lane 4 EtOH, lane 5 EtOH + Fe + taurine, lane 6 EtOH + taurine, and lane 7 CON + taurine
Discussion
Studies have confirmed that the toxicity of ethanol in the liver is due to generation of ROS, depletion of GSH, increased lipid peroxidation, mitochondrial injury, and DNA modification in the liver, where it is oxidized (Nordmann et al. 1992). In our study addition of iron to ethanol-fed rats resulted in marked toxicity. Iron can propagate lipid peroxidation through ferryl–perferryl type oxidants and diverse lipid-derived alkoxyl or peroxyl radicals (Caro and Cederbaum 2001). Addition of iron and ethanol to HepG2 cell lines that overexpress CYP2E1 decreases cell viability and causes apoptosis (Wu and Cederbaum 1996). Iron accumulation is another potential cause of enhanced ROS formation in alcoholic liver disease in humans and associated with increased apoptosis of hepatocytes (Kato et al. 1996). Several investigators have used this combination of treatment at varying concentrations (Stal and Hultcrantz 1993; Tsukamoto et al. 1995; Valerio et al. 1996) to produce fibrosis. The primary reason for the dietary supplementation of iron in addition to intragastric infusion of ethanol was to increase the hepatic non-heme iron higher than normal rather than to produce iron overload. We observed accumulation of iron in both ethanol alone and ethanol plus iron-treated rats.
The ability of ethanol to stimulate mitochondrial ROS production in the hepatocytes is linked to the ability of ethanol to damage the oxidative phosphorylation system (Bailey and Cunningham 2002). Studies have conclusively shown that ethanol alters the concentrations and/or functions of respiratory chain complexes (Hoek 1994). Chronic ethanol consumption decreases the content of iron–sulfur clusters of complex I (Thayer et al. 1980) and levels of cytochrome b in complex III (Cunningham et al. 1990). Chen et al. (2000) observed that liver cytochrome c oxidase is susceptible to oxidative modification and inactivation by the lipid peroxidation products. Prolonged ethanol treatment has been shown to decrease the activity and heme content of cytochrome c oxidase (Spach et al. 1982). Damage to respiratory chain complexes decreases the rate of ATP synthesis which is likely to contribute to the damage observed in hepatocytes. Mitochondrial swelling studies reveal damage to the organelle in ethanol and ethanol plus iron-treated rats. Oxidative stress, membrane damage, and increased membrane pore transition (MPT) are suggested to influence mitochondrial swelling (Cao et al. 2004). Defects in osmoregulation and disturbances in Ca2+ modulation also contribute to mitochondrial swelling.
Chronic exposure to ethanol may alter GSH homeostasis by decreasing the activity of the GSH regulating enzyme, GR (Polavarapu et al. 1998) or by inhibition of synthesis or transport (Fernández-Checa et al. 1998). Also, acetaldehyde, the product of ethanol oxidation can bind to GSH and cause depletion of GSH.
Induction of CYP2E1 by ethanol predisposes to the MPT via ROS production (Cederbaum 2001). The MPT is the regulatable opening of a large, non-specific pore across the outer and inner mitochondrial membrane. Mitochondrial depolarization is followed by cytochrome c release from the mitochondria into cytosol, caspase activation, and apoptosis (Higuchi et al. 2001). High levels of cytoplasmic GSH maintain cytochrome c in a reduced or inactive state. During oxidative stress cytochrome c is released from mitochondria and oxidized. The accumulation of oxidized form of cytochrome c in the cytoplasm can lead to apoptosis (Hancock et al. 2001).
Improved hepatocyte viability, decreased mitochondrial susceptibility to swell, prevention of DNA damage, and reduced apoptosis in the presence of taurine could obviously be attributed to its ability to abrogate oxidative stress. Some studies suggest that taurine functions as a direct antioxidant to scavenge free radicals (Gordon et al. 1986; Chen 1993). Although this direct effect has been questioned by some investigators due to lack of structure–function relationship (Wright et al. 1986; Halliwell and Gutteridge 1990), taurine is still considered to be an antioxidant by its capacity to halt the progression of oxidant-generating reactions. This includes upregulation of antioxidant system, formation of chloramines with hypochlorous acid and replenishment of GSH during oxidative stress. Taurine has been shown to be very effective in reducing lipid peroxidation induced by a variety of toxins such as kainic acid (Baran 2006), carbon tetrachloride (Miyazaki et al. 2005), tamoxifen (Tabassum et al. 2006), and thioacetamide (Balkan et al. 2001).
Besides these functions, taurine can preserve the structural and functional integrity of the cells by acting as a membrane stabilizer, osmoregulator, and a regulator of calcium homeostasis (Milei et al. 1992; Chen 1993). Taurine is cytoprotective to LLC-PK1 cells exposed to dopamine and iron in vitro (Eppler and Dawson 2002). Taurine protects cultured cells under hypoxia and reoxygenation by reducing osmoregulatory dysfunction and improving intracellular calcium concentration (Michalk et al. 1996). Thus the protective effect of taurine on mitochondria might be related to the osmoregulatory effects and through Ca2+ homeostasis. Further studies to measure calcium fluxes and the effects of calcium on swelling and DNA damage are needed to address the action of taurine in the current study. Furthermore, taurine may enhance the anti-inflammatory response by suppressing the activation of nuclear factor-κB (NF-κB) probably by stabilizing the binding protein inhibitor of NF-κB (I-κB). Taurine along with niacin blocks lung fibrosis by inhibiting NF-κB activation and preservation of I-κB (Gurujeyalakshmi et al. 2000). Taurine and its chloramine can modulate the expression and production of pro-inflammatory substances (Schuller-Levis and Park 2004).
Inhibitors of CYP2E1 are suggested to be of therapeutic value in the treatment of alcoholic liver disease (Morimoto et al. 1995). Taurine by conjugation with cholic acid forms taurocholic acid which has been shown to be a potent inhibitor of CYP2E1 (Kerai et al. 1998). In addition, taurine could reduce iron action by chelation (Wu et al. 1999). Taurine has been shown to form complexes with free metal ion species such as Fe2+, Cu2+ with the sulfonic acid group (Trachtman et al. 1992) and works as a chelator at least, in vitro. The benefits may be possibly related to the observation that taurine could scavenge cytotoxic quinones generated as a result of iron-induced oxidation in vivo (Dawson et al. 2000). It is imperative to test whether taurine could perform such a function in vivo. Dietary taurine curtails myocardial iron level, oxidative stress, apoptosis, and fibrosis and replenishes reduced GSH level in iron-overloaded mice. Inhibition of L-type Ca2+ channels that are responsible for iron-uptake and regulation of redox cycling of iron has been proposed as the mechanism for reduction in iron content by taurine by Oudit et al. (2003).
Administration of taurine prevents apoptosis by increasing the level of GSH, a critical determinant of apoptosis and an important factor in regulating the viability of hepatocytes. Taurine stabilizes the GSH-metabolizing enzymes (Hayes et al. 2001), stimulates glucose-6-phosphate dehydrogenase, an enzyme that generates NADPH needed for the regeneration of GSH from GSSG (Tabassum et al. 2006), and modulates GSH levels. Taurine stimulates S-nitrosylation of GSH producing S-nitrosoglutathione, which is more potent than the GSH. S-nitrosylation of cysteine residues by nitrosoglutathione can inactivate caspase-3, thus preventing hepatic cell apoptosis (Chiueh and Rauhala 1999).
Studies in ischemic models have identified the underlying mechanisms of inhibition of apoptosis by taurine. One study observed that taurine suppresses the formation of Apaf-1/caspase-9 apoptosome and interaction of Apaf-1 with caspase-9 (Takatani et al. 2004). Recently, Taranukhin et al. (2008) identified that taurine reduces ischemia induced caspase-8 and caspase-9 expression, the key inducers of apoptosis. Derivatives of taurine also exert downregulating effect on the apoptosis process (Marzioni et al. 2003). Further, taurine prevents hepatic DNA fragmentation and hepatocyte necrosis in acetaminophen-induced hepatotoxicity (Waters et al. 2001)
In our study, taurine, even when ethanol administration was continued, showed its protective effect suggesting that it could abolish the effects of alcohol. Recently, we showed that taurine enhances the metabolism and detoxification of ethanol, and prevents hepatic fibrosis in this model (Devi et al. 2008). In the same study we reported, histological changes induced in this model are reduced by taurine as evidenced by hematoxylin and eosin, and reticulin staining. Although several possible mechanisms seem to explain taurine’s action, additional studies to elucidate the exact mechanisms of action are essential.
Allopathic treatment of alcoholic liver disease is primarily based on supplementation of nutraceuteicals like protein hydrolysate, amino acids, vitamins, and trace elements. Taurine could be an useful adjunct for the treatment of alcoholic liver fibrosis considering the safety profile and the fact that humans have low capacity to synthesize taurine.
References
Anitha Nandhini T, Balakrishnan SD, Anuradha CV (2002) Response of liver antioxidant system to taurine in rats fed high fructose diet. Indian J Exp Biol 40:1016–1019
Bacon BR, Tavill AS, Brittenham GM, Park CH, Recknagel RO (1983) Hepatic lipid peroxidation in vivo in rats with chronic iron overload. J Clin Invest 71:429–439. doi:10.1172/JCI110787
Bailey SM, Cunningham CC (2002) Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol 32:11–16. doi:10.1016/S0891-5849(01)00769-9
Balkan J, Dogru-Abbasoglu S, Kanbağli O, Cevikbas U, Aykaç-Toker G, Uysal M (2001) Taurine has a protective effect against thioacetamide-induced liver cirrhosis by decreasing oxidative stress. Hum Exp Toxicol 20:251–254. doi:10.1191/096032701678227758
Baran H (2006) Alterations of taurine in the brain of chronic kainic acid epilepsy model. Amino Acids 31:303–307. doi:10.1007/s00726-005-0278-6
Brich-Machin M, Jacson S, Klers RS, Turnbull DM (1993) In: Lash LH, Jones DP (eds) Mitochondrial dysfunction. Academic Press, San Diego, CA, pp 51–69
Cao LC, Honeyman TW, Cooney R, Kennington L, Scheid CR, Jonassen JA (2004) Mitochondrial dysfunction is a primary event in renal cell oxalate toxicity. Kidney Int 66:1890–1900. doi:10.1111/j.1523-1755.2004.00963.x
Caro AA, Cederbaum AI (2001) Synergistic toxicity of iron and arachidonic acid in HepG2 cells overexpressing CYP2E1. Mol Pharmacol 60:742–752
Cederbaum AI (2001) Introduction—serial review: alcohol, oxidative stress and cell injury. Free Radic Biol Med 31:1524–1526. doi:10.1016/S0891-5849(01)00741-9
Chen YX (1993) Protective action of taurine on ischemia-reperfusion liver injury in rats and its mechanism. Zhonghua Yi Xue Za Zhi. 73:276–279, 318–319
Chen J, Petersen DR, Schenker S, Henderson GI (2000) Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcohol Clin Exp Res 24:544–552. doi:10.1111/j.1530-0277.2000.tb02023.x
Chiueh CC, Rauhala P (1999) The redox pathway of S-nitrosoglutathione, glutathione and nitric oxide in cell to neuron communications. Free Radic Res 31:41–50. doi:10.1080/10715769900301211
Cunningham CC, Coleman WB, Spach PI (1990) The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol 25:127–136
Dawson R Jr, Baker D, Eppler B, Tang E, Shih D, Hern H, Hu M (2000) Taurine inhibition of metal-stimulated catecholamine oxidation. Neurotox Res 2:1–15
Devi SL, Viswanathan P, Anuradha CV (2008) Taurine enhances the metabolism and detoxification of ethanol and prevents hepatic fibrosis in rats treated with iron and alcohol. Environ Toxicol Pharmacol 27:120–126. doi:10.1016/j.etap.2008.09.004
Dickinson DA, Forman HJ (2002) Cellular glutathione and thiols metabolism. Biochem Pharmacol 64:1019–1026. doi:10.1016/S0006-2952(02)01172-3
Eligini S, Barbieri SS, Cavalca V, Camera M, Brambilla M, De Franceschi M, Tremoli E, Colli S (2005) Diversity and similarity in signaling events leading to rapid Cox-2 induction by tumor necrosis factor-alpha and phorbol ester in human endothelial cells. Cardiovasc Res 65(3):683–693. doi:10.1016/j.cardiores.2004.10.024
Eppler B, Dawson R Jr (2002) Cytoprotective role of taurine in a renal epithelial cell culture model. Biochem Pharmacol 63:1051–1060. doi:10.1016/S0006-2952(02)00843-2
Fernandez-Checa JC, Kaplowitz N (2005) Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol 204:263–273. doi:10.1016/j.taap.2004.10.001
Fernández-Checa JC, Kaplowitz N, García-Ruiz C, Colell A (1998) Mitochondrial glutathione: importance and transport. Semin Liver Dis 18:389–401. doi:10.1055/s-2007-1007172
García-Ruiz C, Morales A, Ballesta A, Rodés J, Kaplowitz N, Fernández-Checa JC (1994) Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest 94:193–201. doi:10.1172/JCI117306
Garlid KD, Beavis AD (1985) Swelling and contraction of the mitochondrial matrix. II. Quantitative application of the light scattering technique to solute transport across the inner membrane. J Biol Chem 260:13434–13441
Gordon RE, Shaked AA, Solano DF (1986) Taurine protects hamster bronchioles from acute NO2-induced alterations. A histologic, ultrastructural, and freeze-fracture study. Am J Pathol 125:585–600
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312. doi:10.1126/science.281.5381.1309
Gurujeyalakshmi G, Wang Y, Giri SN (2000) Taurine and niacin block lung injury and fibrosis by down-regulating bleomycin-induced activation of transcription nuclear factor-kappaB in mice. J Pharmacol Exp Ther 293:82–90
Halliwell B, Gutteridge JM (1990) The antioxidants of human extracellular fluids. Arch Biochem Biophys 280:1–8. doi:10.1016/0003-9861(90)90510-6
Hancock JT, Desikan R, Neill SJ (2001) Does the redox status of cytochrome c act as a fail-safe mechanism in the regulation of programmed cell death? Free Radic Biol Med 31:697–703. doi:10.1016/S0891-5849(01)00646-3
Hayes J, Tipton KF, Bianchi L, Corte LD (2001) Complexities in the neurotoxic actions of 6-hydroxydopamine in relation to the cytoprotective properties of taurine. Brain Res Bull 55:239–245. doi:10.1016/S0361-9230(01)00507-X
Higuchi H, Adachi M, Miura S, Gores GJ, Ishii H (2001) The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology 34:320–328. doi:10.1053/jhep.2001.26380
Hoek JB (1994) Mitochondrial energy metabolism in chronic alcoholism. Curr Top Bioenerg 17:197–241
Horn HD, Burn FH (1978) In: Bergmeyer HU (ed) Methods in enzymology. Academic Press, New York, pp 877
Huxtable RJ (1992) Physiological actions of taurine. Physiol Rev 72:101–163
Hwang DF, Wang LC (2001) Effect of taurine on toxicity of cadmium in rats. Toxicology 167:173–180. doi:10.1016/S0300-483X(01)00472-3
Jiang ZY, Hunt JV, Wolff SP (1992) Ferrous ion oxidation in the presence of xylenol orange for detection of lipid hydroperoxide in low density lipoprotein. Anal Biochem 202:384–389. doi:10.1016/0003-2697(92)90122-N
Kato J, Kobune M, Kohgo Y, Sugawara N, Hisai H, Nakamura T, Sakamaki S, Sawada N, Niitsu Y (1996) Hepatic iron deprivation prevents spontaneous development of fulminant hepatitis and liver cancer in Long-Evans Cinnamon rats. J Clin Invest 98:923–929. doi:10.1172/JCI118875
Kerai MD, Waterfield CJ, Kenyon SH, Asker DS, Timbrell JA (1998) Taurine protective properties against ethanol-induced hepatic steatosis and lipid peroxidation during chronic ethanol consumption in rats. Amino Acids 15:53–76. doi:10.1007/BF01345280
Lieber CS (1997) Cytochrome P-4502E1: its physiological and pathological role. Physiol Rev 77:517–544
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275
Marzioni M, LeSage GD, Glaser S, Patel T, Marienfeld C, Ueno Y, Francis H, Alvaro D, Tadlock L, Benedetti A, Marucci L, Baiocchi L, Phinizy JL, Alpini G (2003) Taurocholate prevents the loss of intrahepatic bile ducts due to vagotomy in bile duct-ligated rats. Am J Physiol Gastrointest Liver Physiol 284:G837–G852
Michalk DV, Wingenfeld P, Licht C, Ugur T, Siar LF (1996) The mechanisms of taurine mediated protection against cell damage induced by hypoxia and reoxygenation. Adv Exp Med Biol 403:223–232
Milei J, Ferreira R, Llesuy S, Forcada P, Covarrubias J, Boveris A (1992) Reduction of reperfusion injury with preoperative rapid intravenous infusion of taurine during myocardial revascularization. Am Heart J 123:339–345. doi:10.1016/0002-8703(92)90644-B
Miyazaki T, Karube M, Matsuzaki Y, Ikegami T, Doy M, Tanaka N, Bouscarel B (2005) Taurine inhibits oxidative damage and prevents fibrosis in carbon tetrachloride-induced hepatic fibrosis. J Hepatol 43:117–125. doi:10.1016/j.jhep.2005.01.033
Morimoto M, Hagbjörk AL, Wan YJ, Fu PC, Clot P, Albano E, Ingelman-Sundberg M, French SW (1995) Modulation of experimental alcohol-induced liver disease by cytochrome P450 2E1 inhibitors. Hepatology 21:1610–1617
Niehaus WG, Samuelsson B (1968) Formation of malonaldehyde from phospholipid arachidonate during microsomal lipid peroxidation. Eur J Biochem 6:126–130. doi:10.1111/j.1432-1033.1968.tb00428.x
Nordmann R, Ribiere C, Rouach H (1992) Implication of free radical mechanisms in ethanol-induced cellular injury. Free Radic Biol Med 12:219–240
Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, Yazdanpanah M, Wilson GJ, Schwartz A, Liu PP, Backx PH (2003) L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med 9:1187–1194. doi:10.1038/nm920
Polavarapu R, Spitz DR, Sim JE, Follansbee MH, Oberley LW, Rahemtulla A, Nanji AA (1998) Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology 27:1317–1323. doi:10.1002/hep.510270518
Pushpakiran G, Mahalakshmi K, Anuradha CV (2004) Taurine restores ethanol-induced depletion of antioxidants and attenuates oxidative stress in rat tissues. Amino Acids 27:91–96. doi:10.1007/s00726-004-0066-8
Pushpakiran G, Mahalakshmi K, Viswanathan P, Anuradha CV (2005) Taurine prevents ethanol-induced alterations in lipids and ATPases in rat tissues. Pharmacol Rep 57:578–587
Schaffer S, Azuma J, Takahashi K, Mozaffari M (2003) Why is taurine cytoprotective? Adv Exp Med Biol 526:307–321
Schuller-Levis GB, Park E (2004) Taurine and its chloramine: modulators of immunity. Neurochem Res 29:117–126. doi:10.1023/B:NERE.0000010440.37629.17
Seglen PO (1976) Preparation of isolated rat liver cells. Methods Cell Biol 13:29–83. doi:10.1016/S0091-679X(08)61797-5
Slater EC, Bonner WD (1952) Effect of fluoride on succinate oxidase system. J Biochem 52:185–196
Spach PI, Bottenus RE, Cunningham CC (1982) Control of adenine nucleotide metabolism in hepatic mitochondria from rats with ethanol-induced fatty liver. Biochem J 202:445–452
Stal P, Hultcrantz R (1993) Iron increases ethanol toxicity in rat liver. J Hepatol 17:108–115. doi:10.1016/S0168-8278(05)80530-6
Tabassum H, Rehman H, Banerjee BD, Raisuddin S, Parvez S (2006) Attenuation of tamoxifen-induced hepatotoxicity by taurine in mice. Clin Chim Acta 370:129–136. doi:10.1016/j.cca.2006.02.006
Takatani T, Takahashi K, Uozumi Y, Shikata E, Yamamoto Y, Ito T, Matsuda T, Schaffer SW, Fujio Y, Azuma J (2004) Taurine inhibits apoptosis by preventing formation of the Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol 287:C949–C953. doi:10.1152/ajpcell.00042.2004
Taranukhin AG, Taranukhina EY, Saransaari P, Djatchkova IM, Pelto-Huikko M, Oja SS (2008) Taurine reduces caspase-8 and caspase-9 expression induced by ischemia in the mouse hypothalamic nuclei. Amino Acids 34:169–174. doi:10.1007/s00726-006-0405-z
Thayer WS, Ohnishi T, Rubin E (1980) Characterization of iron–sulfur clusters in rat liver submitochondrial particles by electron paramagnetic resonance spectroscopy. Alterations produced by chronic ethanol consumption. Biochim Biophys Acta 591:22–36. doi:10.1016/0005-2728(80)90217-0
Tietze F (1969) Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione. Anal Biochem 27:502–522
Trachtman H, Del Pizzo R, Futterweit S, Levine D, Rao PS, Valderrama E, Sturman JA (1992) Taurine attenuates renal disease in chronic puromycin aminonucleoside nephropathy. Am J Physiol 262:117–123
Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, Brittenham GM (1995) Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest 96:620–630. doi:10.1172/JCI118077
Uysal M, Ozdemirler G, Kutalp G, Oz H (1989) Mitochondrial and microsomal lipid peroxidation in rat liver after acute acetaldehyde and ethanol intoxication. J Appl Toxicol 9:155–158. doi:10.1002/jat.2550090304
Valerio LG Jr, Parks T, Petersen DR (1996) Alcohol mediates increases in hepatic and serum nonheme iron stores in a rat model for alcohol-induced liver injury. Alcohol Clin Exp Res 20:1352–1361. doi:10.1111/j.1530-0277.1996.tb01134.x
Waters E, Wang JH, Redmond HP, Wu QD, Kay E, Bouchier-Hayes D (2001) Role of taurine in preventing acetaminophen-induced hepatic injury in the rat. Am J Physiol Gastrointest Liver Physiol 280:G1274–G1279
Wharton DC, Tzagoloff A (1967) Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 10:245–250. doi:10.1016/0076-6879(67)10048-7
Wright CE, Tallan HH, Lin YY, Gaull GE (1986) Taurine: biological update. Annu Rev Biochem 55:427–453. doi:10.1146/annurev.bi.55.070186.002235
Wu D, Cederbaum AI (1996) Ethanol cytotoxicity to a transfected HepG2 cell line expressing human cytochrome P4502E1. J Biol Chem 271:23914–23919. doi:10.1074/jbc.271.39.23914
Wu QD, Wang JH, Fennessy F, Redmond HP, Bouchier-Hayes D (1999) Taurine prevents high-glucose-induced human vascular endothelial cell apoptosis. Am J Physiol 277:C1229–C1238
Yacoub LK, Fogt F, Griniuviene B, Nanji AA (1995) Apoptosis and bcl-2 protein expression in experimental alcoholic liver disease in the rat. Alcohol Clin Exp Res 19:854–859. doi:10.1111/j.1530-0277.1995.tb00958.x
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lakshmi Devi, S., Anuradha, C.V. Mitochondrial damage, cytotoxicity and apoptosis in iron-potentiated alcoholic liver fibrosis: amelioration by taurine. Amino Acids 38, 869–879 (2010). https://doi.org/10.1007/s00726-009-0293-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0293-0