
Abstract
Apoptosis proteins have a central role in the development and the homeostasis of an organism. These proteins are very important for understanding the mechanism of programmed cell death. The function of an apoptosis protein is closely related to its subcellular location. It is crucial to develop powerful tools to predict apoptosis protein locations for rapidly increasing gap between the number of known structural proteins and the number of known sequences in protein databank. In this study, amino acids pair compositions with different spaces are used to construct feature sets for representing sample of protein feature selection approach based on binary particle swarm optimization, which is applied to extract effective feature. Ensemble classifier is used as prediction engine, of which the basic classifier is the fuzzy K-nearest neighbor. Each basic classifier is trained with different feature sets. Two datasets often used in prior works are selected to validate the performance of proposed approach. The results obtained by jackknife test are quite encouraging, indicating that the proposed method might become a potentially useful tool for subcellular location of apoptosis protein, or at least can play a complimentary role to the existing methods in the relevant areas. The supplement information and software written in Matlab are available by contacting the corresponding author.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Computational approaches, such as structural bioinformatics (Argos et al. 1982; Chou 2004a, b, c, d, 2005a), molecular docking (Chou et al. 2003; Gao et al. 2007; Li et al. 2007; Wang et al. 2008; Zhang et al. 2006a, b, c; Zheng et al. 2007), molecular packing (Chou et al. 1984, 1988), pharmacophore modeling (Chou et al. 2006; Sirois et al. 2004), Mote Carlo simulated approach (Chou 1992), diffusion-controlled reaction simulation (Chou and Jiang 1974; Chou and Zhou 1982; Li and Chou 1976), bio-macromolecular internal collective motion simulation (Chou 1988), QSAR (Dea-Ayuela et al. 2008; Du et al. 2005, 2008a, b; Gonzalez-Diaz et al. 2006, 2008; Prado-Prado et al. 2008), protein subcellular location prediction (Chou and Shen 2006a, b, 2007a, c, 2008a; Shi et al. 2008), identification of membrane proteins and their types (Chou and Shen 2007b), identification of enzymes and their functional classes (Shen and Chou 2007a), identification of GPCR and their types (Chou 2005b; Chou and Elrod 2002), identification of proteases and their types (Chou and Shen 2008b), protein cleavage site prediction (Chou 1993, 1996; Shen and Chou 2008), and signal peptide prediction (Chou and Shen 2007d; Shen and Chou 2007b) can timely provide very useful information and insights for both basic research and drug design and hence are widely welcome by science community. The present study is attempted to develop a computational approach for predicting the subcellular localization of apoptosis proteins in hope to stimulate the development of the relevant areas (Emanuelsson et al. 2007; Fauchere et al. 1988; Janin 1979; Janin and Wodak 1978).
Apoptosis is a form of cell death which plays a central role in normal tissue homeostasis by regulating a balance between cell proliferation and death (Chou et al. 1997, 1999, 2000; Chou 2004a, b, c, d, 2005a, b, c). Cells undergoing apoptosis usually exhibit a characteristic morphology, including fragmentation of the cell into membrane-bound apoptotic bodies, nuclear and cytoplasm condensation and hemolytic cleavage of the DNA into small oligonucleosomal fragments (Kerr et al. 1972; Steller 1995). Unregulated excessive apoptosis may cause various degenerative and autoimmune diseases. Conversely, an inappropriately low rate of apoptosis may promotes survival and accumulation of abnormal cells that can give rise to tumor formation and prolonged autoimmune stimulation such as in cancers and Graves disease (Peter et al. 1997).The study on apoptosis proteins can help us to understand the mechanism of apoptosis and provide many targets for therapeutic intervention (Cosic 1994; Du and Li 2006; Hong et al. 1999; Hopp and Woods 1981; Huang and Shi 2005; Chou 2000, Chou 2004a, b, c, d, 2005a, b, c).
The function of a protein is closely correlated with its subcellular location (Cai and Chou 2003; Cai et al. 2003; Chou 2002; Chou and Cai 2002, 2004, 2005; Chou and Shen 2006a, b, c; Shen et al. 2007b; Shen et al. 2005; Shen and Chou 2007; Chou and Elrod 1999; Chou 2000, 2001; Feng 2002). Thus, the knowledge of apoptosis proteins subcellular location will help to understand the apoptosis mechanism and functions of proteins (Schulz et al. 1999; Reed and Paternostro 1999). The knowledge of apoptosis proteins function is very important for understanding the mechanism of programmed cell death. The malfunction of apoptosis or cell death will lead to some formidable diseases, such as cancer (Adams and Cory 1998; Evan and Littlewood 1998), autoimmune diseases, ischemic damage, or neurodegenerative disease (Schulz et al. 1999). With the rapid increasing of the number of unknown function protein sequences in protein databank, it is crucial to develop fast and powerful computational tools and algorithms to predict apoptosis proteins subcellular location directly from their amino acid sequences.
Several prediction algorithms have been reported for subcellular location of apoptosis protein. Zhou and Doctor (2003) have predicted four kinds of subcellular locations by using amino acid composition (AAC) representing sample of protein, and covariant discriminate algorithm of Chou (1995) as prediction engine. They obtained overall accuracy 72.5% by jackknife test. Bulashevska and Eils (2006) achieved accuracies 85.7 and 89.9% using single Bayesian classifier and hierarchical ensemble classifier, respectively. Zhang et al. (2006b) developed a new encoding method with grouped weight for protein sequence. Meanwhile, they constructed a larger dataset with 225 apoptosis protein belonged to four subcellular locations. A prediction algorithm of dual-layer support vector machine has been developed (Zhou et al. 2008). Chen and Li (2007a, b) have developed two prediction approaches based on increment of diversity (ID) and increment of diversity with support vector machine (ID_SVM), which are validated on a new dataset covering six subcellular compartments and 317 apoptosis proteins.
Compare to lots of research on protein subcellular location (Chou and Shen 2007b), the studies on apoptosis protein subcellular location are limited. It is mainly due to the flexibility of the apoptosis proteins distribution and the limited of apoptosis proteins annotated. In this study, we propose a new prediction approach based on ensemble classifier and feature selection for prediction of apoptosis protein subcellular location based on the analysis above mentioned. A new kind of ensemble classifier is introduced as prediction engine. The methods of ensemble classifier, which has the capability of reducing the variance caused by the peculiarities of a single training set and hence be able to learn a more expressive concept in classification than a single classifier, are proposed in various attributes of protein science (Shen and Chou 2006a, b, 2007a; Shen et al. 2007a; Kedarisetti et al. 2006; Chou and Shen 2006a, 2006b, 2007a). The basic classifier is fuzzy K-nearest neighbor (FKNN) (Keller et al. 1985) classifier, which is a simple and powerful classifier often used in identifying various protein attributes (Huang and Li 2004; Shen et al. 2006; Huang et al. 2006). For each basic classifier within ensemble classifier, the input data is k-spaced amino acid pair’s composition after feature selection. The test results obtained by jackknife test indicate that the proposed method might be a useful tool for subcellular location of apoptosis protein, or at least can play a complimentary role to the existing methods in the relevant areas.
Materials and methods
Datasets
Two datasets constructed by the previous investigators are used to examine the power of the new method. The dataset CL317 is a larger one with 317 apoptosis proteins constructed by Chen and Li (2007a), which has 112 cytoplasmic proteins, 55 membrane proteins, 34 mitochondrial proteins, 17 secreted proteins, 52 nuclear proteins, and 47 endoplasmic reticulum proteins. The dataset ZW225 with 225 apoptosis proteins in the work (Zhang et al. 2006b) includes four subcellular locations with 41 nuclear proteins, 70 cytoplasmic proteins, 25 mitochondrial proteins, and 89 membrane proteins.
K-spaced amino acid pairs
As mentioned in prior works, amino acids (AA) composition vector of protein sequence is a simple sequence representation that is widely used in prediction of various structural aspects. Given 20 alphabetically ordered (A, C,D, E, F, G, H, I, K, L, M, N, P, Q, R, S, T, V, W, Y) AA, which are denoted as A 1 , A 2 ,…, A 19 , and A 20 , and the number of occurrences of A i in the sequence that is denoted as x i , the composition vector is defined as (x 1/L, x 2/L,…, x i /L), where L is the length of the sequence (Chen et al. 2006b, 2007b; Kawashima et al. 1999; Nakashima and Nishikawa 1994; Park and Kanehisa 2003; Pincus 1991; Richman and Moorman 2000; Shi et al. 2007; Tanford 1962; Zimmerman et al. 1968). However, the composition vector is insufficient to represent a sequence, since it only counts the frequencies of individual AAs. At the same time, frequencies of AA pairs (dipeptides) provide more information since they reflect interaction between local (with respect to the sequence) AA pairs. Based on the frequency of collocation of AA pairs in the sequence, all dipeptides in the sequence can be counted. Since there are 400 possible dipeptides (AA, AC, AD,…,YY), a feature vector of that size is used to represent occurrence of these pairs in the sequence. Each AA pairs occurrence rate is (n 1/(L − 1), n 2/(L − 1),…, n i /(L − 1)). Since short-range interactions between AAs, rather than only interactions between immediately adjacent AAs, have impact of folding, the proposed representation also considers collocated pairs of AAs, i.e., the AA pairs that are separated by p other AAs (e.g., the AA pairs form is AA 1 A 2 …A p A, where A 1 A 2 …A p are other AA). In summary, these pairs can be understood as the dipeptides with gaps. For each value of p, there are 400 corresponding feature values. At the same time, each AA pairs occurrence rate is reduced to (n 1/(L − p − 1), n 2/(L − p − 1),…, n i /(L − p − 1)). Collocated pairs for p = 0, 1,…, 20 are considered for the reason that the distance of AA in motif database PROSITE is up to 20 (Chen et al. 2007a; Falquet et al. 2002). As a result, we propose representation that includes total of 400(20 + 1) + 20 = 8,420 features.
Binary particle swarm optimization
Particle swarm optimization (PSO) is a population-based stochastic optimization technique, which was developed by Kennedy and Eberhart (1995). It is one of the evolutionary optimization methods inspired by nature which include evolutionary strategy, evolutionary programming, genetic algorithm and genetic programming. PSO is distinctly different from other evolutionary-type methods in that it does not use the filtering operation (such as crossover and/or mutation) and the members of the entire population are maintained through the search procedure (Kennedy et al. 2001).
In the PSO algorithm, every solution is a bird of the flock and is referred to as a particle: in this framework the birds, besides having individual intelligence, also develop some social behavior and coordinate their movement towards a destination.
Initially, the process starts from a swarm of particles, in which each of them contains a solution to the hydraulic problem that is generated randomly, and then one searches the optimal solution by iteration. The ith particle is associated with a position in an s-dimensional space, where M is the number of variables involved in the problem; the values of the M variables which determine the position of the particle represent a possible solution of the optimization problem. Each particle i is completely determined by three vectors: its current position X i, and its velocity V i as follows:
This algorithm simulates a flock of birds which communicate during flight. Each bird looks at a specific direction (its best ever attained position), and later, when they communicate among themselves, the bird which is in the best position is identified. With coordination, each bird moves also towards the best bird using a velocity which depends on its present velocity. Thus, each bird examines the search space from its current local position, and this process repeats until the bird possibly reaches the desired position. Note that this process involves as much individual intelligence as social interactivity; the birds learn through their own experience (local search) and the experience of their peers (global search).
In each cycle, one identifies the particle which has the best instantaneous solution to the problem; the position of this particle subsequently enters into the computation of the new position for each of the particles in the flock. This calculation is carried out according to
Here, rand() represents a function which creates random numbers between 0 and 1 (two independent random numbers enter Eq. 4); pbest k id represents the best position of each particle i reached in kth cycle whereas gbest represents the best result of global search. C 1 and C 2 are two positive constants which are called learning factors or rates which are usually set to 2.
PSO was originally introduced as an optimization technique for real-number spaces. However, many optimization problems occur in a space featuring discrete, qualitative distinctions between variables and between levels of variables. Kennedy and Eberhart introduced binary PSO (BPSO), which can be applied to discrete binary variables. In a binary space, a particle may move to near corners of a hypercube by flipping various numbers of bits; thus, the overall particle velocity may be described by the number of bits changed per iteration (Kennedy and Eberhart 1997). In BPSO, each particle position X i is set to 1 or 0, but the flight velocity V i are not limited. In our paper, BPSO is used as feature selection algorithm. All the AA pairs feature of apoptosis proteins above mentioned compose of particle space. If the ith feature is selected, then X i = 1; if not, X i = 0. The fitness function of the feature selection algorithm is formulated by Eq. 5.
In Eq. 5, Ac represents the accuracy of Jackknife test (Chou and Zhang 1995) on training dataset, nNewFeature represents the number of newly features selected, nAllFeature represents the number of all features, and k is a parameter represents the fixed ratio of feature selected in the algorithm. In our paper, k = 1.
Based on the velocity of particles in BPSO calculated by Eq. 4, each particle’s new position X k+1 id can be get as follows:
where a is a parameter represents updating of particles, in our research a = 1.
Ensemble classifier
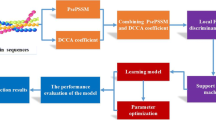
The framework of the ensemble classifier is illustrated in Fig. 1. The basic classifier is FKNN classifier which is trained on the k-spaced amino acid pair’s composition after feature selection. Combining a set of basic classifiers, the ensemble classifier is formulated by
where C denotes the ensemble classifier, C i {BiAAC(p = i)}, i = 0, 1, , n, represent the basic classifiers trained by proteins based on the feature selection results of p-spaced amino acid pairs composition. The symbol ⊕ is the combination operator. Here, the basic classifier is the FKNN classifier (Keller et al. 1985), which combines the fuzzy set theory with KNN algorithm. The detailed algorithm description of the FKNN can be found in the work (Huang and Li 2004; Shen et al. 2006; Zheng et al. 2007). The output of each basic classifier is the fuzzy membership value of subcellular location of apoptosis protein. A fuzzy membership matrix can be formulated as Eq. 8
where c is the number of subcellular location and n is the number of k-spaced amino acid pair.

The flowchart of prediction approach and framework of ensemble classifier
Through fusing the output of each basic classifier, the fuzzy membership value of output of ensemble classifier can be obtained.
where u i = (m 1 i (x), m 2 i (x),…, m n i (x)), i = 1, 2,…, c, “comb” express the rule of fusion. The final result is the maximum of f i in Eq. 10.
Fuzzy K-nearest neighbor classifier
Combining the fuzzy set theory with KNN algorithm, Keller has proposed a new method named as FKNN classifier algorithm (Keller et al. 1985). The fuzzy membership of a sample of protein is assigned to different subcellular location according to the formulation as below:
where k is the number of nearest neighbors, u i (p) is the membership value of a protein sample to structural class i. m is the fuzzy parameter, which determines the weight of distance of each neighbor to membership value. ||p − p (j)|| is the distance between the test protein sample and it nearest neighbor samples, various distance functions can be chosen, Here, we use Euclidean distance. u i (p (j)) is the membership value of the jth nearest neighbor to ith subcellular location. It is assigned in crispest way, which is illuminated as below.
When all memberships of each subcellular location are calculated, the test protein sample is assigned to the class with highest membership value. As the prior work we did, it is a useful prediction engine (Zhang et al. 2006a, b, c, 2008). For the reason that p = 0, 1,…, 20 in feature selection in our research, 21 FKNN are selected as basic classifiers of ensemble classifier.
Performance measurement
To measure the quality of apoptosis protein subcellular locations prediction, it is convenient to introduce an accuracy matrix [M ii ] of size c × c (c is the number of compartments to be predicted). The element M ii of the accuracy matrix is the number of proteins to be predicted in subcellular location j, which are actually in subcellular location i.
Three indexes are applied to evaluate the prediction accuracy, which are sensitivity (S n ), specialty (S p ), and Matthew’s correlation coefficients (MCC).
S n represents the accuracy, and S p represents the reliability in prediction. MCC is a single parameter characterizing the matching degree between the observed and predicted structural classes.
Results and discussion
In statistical prediction, the following three cross-validation tests are often used to examine the power of a predictor: independent dataset test, sub-sampling (such fivefold or tenfold sub-sampling) test, and jackknife test (Chou and Zhang 1995; Cai et al. 2001; Zhou and Assa-Munt 2001; Zhou 1998). Of these three, however, the jackknife test is thought the most rigorous and objective that can always yield a unique result for a given benchmark dataset, as elucidated in (Zhou and Cai 2006; Chou and Shen 2008a) and demonstrated by Eq. 50 of (Chou and Shen 2007c), and hence has been used by more and more investigators (e.g., Chen et al. 2006a, b; Gao et al. 2005a, b; Liu et al. 2005a; Liu et al. 2005b; Chou and Shen 2006a, b, 2007a; Xiao et al. 2005; 2006a, b; Lin and Li 2007a, b; Zhang et al. 2006a, b; Zheng et al. 2007) in examining the power of various prediction methods.
Firstly, the dataset CL317 (Chen and Li 2007a) is applied to validate our research approach. The dimension of protein features and jackknife test result are showed in Table 1.
From Table 1, we can see that the features dimension of different k-space has been reduced, while the jackknife accuracy of each basic classifier reasonably increases after feature selection. The reason for that is using BPSO as the feature selection method can reduce the redundancy features efficiently.
After ensemble the 21 FKNN classifiers as prediction engine, the jackknife results on CL137 dataset are listed in Table 2.
As shown in Table 2, the overall accuracy of jackknife test is 91.5% by using ensemble classifier with 21 trained FKNN weak classifiers, 1–3% higher than using only one FKNN classifier (Table 1). The reason is listed as follows: the ensemble classifier, which has the capability of reducing the imbalance caused by the peculiarities of a single training set and hence be able to learn a more expressive concept in classification than a single classifier, are proposed in various attributes of protein science (Shen and Chou 2006a, b, 2007a). From the Table 2 we also can find two results: firstly, the result of our approach is obviously higher than ID (Chen and Li 2007a) and ID_SVM (Chen and Li 2007b) in the same dataset. The reason is our protein features after feature selection are more effective than that of two methods. From Table 2, we can see the jackknife results are obviously higher in Cytoplasmic, Nuclear proteins and endoplasmic reticulum location than ID and ID_SVM methods.
In order to validate the performance of the proposed approach further, the dataset ZW225 (Zhang et al. 2006b) is adopted. The jackknife results are shown in Table 3.
As shown in Table 3, the overall prediction accuracy Ac(%) of this study is the highest both in total accuracy and success rate in each subcellular compartment. What is more, from the Tables 2 and 3 we can see the desirable values of S n , S p , MCC, which also verify the objective of jackknife test.
Conclusions
In this paper, binary particle swarm optimization (BPSO) is applied to extract effective feature, and AA pair compositions with different spaces are used to construct feature sets for protein feature selection. In order to increase prediction accuracy, ensemble classifier is applied as prediction engine, of 21 classifier is the FKNN (fuzzy K-nearest neighbor) trained with different feature sets. Two datasets CL317 and ZW225 are selected to validate the performance of proposed approach, the jackknife result are 91.5 and 88.0%, respectively, which both are the better than other methods. The results indicate that the proposed method will be a potentially useful tool for subcellular location of apoptosis protein.
References
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326
Argos P, Rao JK, Hargrave PA (1982) Structural prediction of membrane-bound proteins. Eur J Biochem 128:565–575
Bulashevska A, Eils R (2006) Predicting protein subcellular locations using hierarchical ensemble of Bayesian classifiers based on Markov chains. BMC Bioinform 7:298–310
Cai YD, Chou KC (2003) Nearest neighbour algorithm for predicting protein subcellular location by combining functional domain composition and pseudo-amino acid composition. Biochem Biophys Res Commun 305:407–411
Cai YD, Liu XJ, Xu XB, Zhou GP (2001) Support vector machines for predicting structural class. BMC Bioinform 2:3
Cai YD, Zhou GP, Chou KC (2003) Support vector machines for predicting membrane protein types by using functional domain composition. Biophys J 84:3257–3263
Chen YL, Li QZ (2007a) Prediction of the subcellular location of apoptosis proteins. J Theor Biol 245:775–783
Chen YL, Li QZ (2007b) Prediction of apoptosis protein subcellular location using improved hybrid approach and pseudo-amino acid composition. J Theor Biol 248(2):377–381. doi:10.1016/j.jtbi.2007.05.019
Chen C, Zhou X, Tian YX, Zou XY, Cai PX (2006a) Predicting protein structural class with pseudo-amino acid composition and support vector machine fusion network. Anal Biochem 357:116–121
Chen C, Tian YX, Zou XY, Cai PX, Mo JY (2006b) Using pseudo-amino acid composition and support vector machine to predict protein structural class. J Theor Biol 243:444–448
Chen K, Kurgan LA, Rahbari M (2007a) Prediction of protein crystallization using collocation of amino acid pairs. Biochem Biophys Res Commun 355:764–769
Chen K, Kurgan LA, Ruan JH (2007b) Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs. BMC Struct Biol 7:25
Chou KC (1988) Review: low-frequency collective motion in biomacromolecules and its biological functions. Bio Chem 30:3–48
Chou KC (1992) Energy-optimized structure of antifreeze protein and its binding mechanism. J Mol Biol 223:509–517
Chou KC (1993) A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. J Biol Chem 268:16938–16948
Chou KC (1995) A novel approach to predicting protein structural classes in a (20–1)-D amino acid composition space. Proteins 21:319–344
Chou KC (1996) Review: prediction of HIV protease cleavage sites in proteins. Anal Biochem 233:1–14
Chou KC (2000) Review: prediction of protein structural classes and subcellular locations. Curr Protein Pept Sci 1:171–208
Chou KC (2001) Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins Struct Funct Genet 43(3):246–255
Chou KC (2002) A new branch of proteomics: prediction of protein cellular attributes. In: Weinrer PW, Lu Q (eds) Gene cloning and expression technologies. Eaton Publishing, Westborough, pp 57–70
Chou KC (2004a) Review: structural bioinformatics and its impact to biomedical science. Curr Med Chem 11:2105–2134
Chou KC (2004b) Insights from modelling the 3D structure of the extracellular domain of alpha7 nicotinic acetylcholine receptor. Biochem Biophys Res Commun 319:433–438
Chou KC (2004c) Modelling extracellular domains of GABA-A receptors: subtypes 1, 2, 3, and 5. Biochem Biophys Res Commun 316:636–642
Chou KC (2004d) Molecular therapeutic target for type-2 diabetes. J Proteome Res 3:1284–1288
Chou KC (2005a) Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21:10–19
Chou KC (2005b) Coupling interaction between thromboxane A2 receptor and alpha-13 subunit of guanine nucleotide-binding protein. J Proteome Res 4:1681–1686
Chou KC (2005c) Prediction of G-protein-coupled receptor classes. J Proteome Res 4:1413–1418
Chou KC, Cai YD (2002) Using functional domain composition and support vector machines for prediction of protein subcellular location. J Biol Chem 277:45765–45769
Chou KC, Cai YD (2004) Prediction of protein subcellular locations by GO-FunD-PseAA predictor. Biochem Biophys Res Commun 320:1236–1239
Chou KC, Cai YD (2005) Predicting protein localization in budding yeast. Bioinformatics 21:944–950
Chou KC, Elrod DW (1999) Prediction of membrane protein types and subcellular locations. Proteins: Struct Funct Genet 34:137–153
Chou KC, Elrod DW (2002) Bioinformatical analysis of G-protein-coupled receptors. J Proteome Res 1:429–433
Chou KC, Jiang SP (1974) Studies on the rate of diffusion-controlled reactions of enzymes. Sci Sinica 17:664–680
Chou KC, Shen HB (2006a) Hum-PLoc: a novel ensemble classifier for predicting human protein subcellular localization. Biochem Biophys Res Commun 347:150–157
Chou KC, Shen HB (2006b) Predicting eukaryotic protein subcellular location by fusing optimized evidence-theoretic K-nearest neighbor classifiers. J Proteome Res 5:1888–1897
Chou KC, Shen HB (2006c) Predicting protein subcellular location by fusing multiple classifiers. J Cell Biochem 99:517–527
Chou KC, Shen HB (2007a) Euk-mPLoc: a fusion classifier for large-scale eukaryotic protein subcellular location prediction by incorporating multiple sites. J Proteome Res 6:1728–1734
Chou KC, Shen HB (2007b) MemType-2L: a Web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM. Biochem Biophys Res Comm 360:339–345
Chou KC, Shen HB (2007c) Review: recent progresses in protein subcellular location prediction. Anal Biochem 370:1–16
Chou KC, Shen HB (2007d) Signal-CF: a subsite-coupled and window-fusing approach for predicting signal peptides. Biochem Biophys Res Comm 357:633–640
Chou KC, Shen HB (2008a) Cell-PLoc: a package of web-servers for predicting subcellular localization of proteins in various organisms. Nat Protoc 3:153–162
Chou KC, Shen HB (2008b) ProtIdent: a web server for identifying proteases and their types by fusing functional domain and sequential evolution information. Biochem Biophys Res Comm 376(2):321–325. doi:10.1016/j.bbrc.2008.08.125
Chou KC, Zhang CT (1995) Review: prediction of protein structural classes. Crit Rev Biochem Mol Bio 30:275–349
Chou KC, Zhou GP (1982) Role of the protein outside active site on the diffusion-controlled reaction of enzyme. J Am Chem Soc 104:1409–1413
Chou KC, Nemethy G, Scheraga HA (1984) Energetic approach to packing of a-helices: 2. General treatment of nonequivalent and nonregular helices. J American Chem Soc 106:3161–3170
Chou KC, Maggiora GM, Nemethy G, Scheraga HA (1988) Energetics of the structure of the four-alpha-helix bundle in proteins. Proc Natl Acad Sci U S A 85:4295–4299
Chou KC, Zhang TC, Maggiora MG (1997) Disposition of amphiphilic helices in heteropolar environments. Proteins 28:99–108
Chou JJ, Li H, Salvessen GS, Yuan J, Wagner G (1999) Solution structure of BID, an intracellular amplifier of apoptotic signalling. Cell 96:615–624
Chou KC, Tomasselli AG, Heinrikson RL (2000) Prediction of the tertiary structure of a caspase-9/inhibitor complex. FEBS Lett 470:249–256
Chou KC, Wei DQ, Zhong WZ (2003) Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS (Erratum: ibid., 2003, Vol. 310, 675). Biochem Biophys Res Comm 308:148–151
Chou KC, Wei DQ, Du QS, Sirois S, Zhong WZ (2006) Review: progress in computational approach to drug development against SARS. Curr Med Chem 13:3263–3270
Cosic I (1994) Macromolecular bioactivity: is it resonant interaction between macromolecules?—theory and applications. IEEE Trans Biomed Eng 41:1101–1114
Dea-Ayuela MA, Perez-Castillo Y, Meneses-Marcel A, Ubeira FM, Bolas-Fernandez F, Chou KC, Gonzalez-Diaz H (2008) HP-Lattice QSAR for dynein proteins: Experimental proteomics (2D-electrophoresis, mass spectrometry) and theoretic study of a Leishmania infantum sequence. Bioorg Med Chem 16:7770–7776
Du PF, Li YD (2006) Prediction of protein submitochondria locations by hybridizing pseudo-amino acid composition with various physicochemical features of segmented sequence. BMC Bioinform 7:518–526
Du QS, Mezey PG, Chou KC (2005) Heuristic molecular lipophilicity potential (HMLP): a 2D-QSAR study to LADH of molecular family pyrazole and derivatives. J Comput Chem 26:461–470
Du QS, Huang RB, Chou KC (2008a) Review: recent advances in QSAR and their applications in predicting the activities of chemical molecules, peptides and proteins for drug design. Curr Protein Pept Sci 9:248–259
Du QS, Huang RB, Wei YT, Du LQ, Chou KC (2008b) Multiple field three dimensional quantitative structure-activity relationship (MF-3D-QSAR). J Comput Chem 29:211–219
Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2:953–971
Evan G, Littlewood T (1998) A matter of life and cell death. Science 281:1317–1322
Falquet L, Pagni M, Bucher P, Hulo N, Sigrist CJ, Hofmann K, Bairoch A (2002) The PROSITE database, its status in 2002. Nucleic Acids Res 30:235–238
Fauchere JL, Charton M, Kier LB, Verloop A, Pliska V (1988) Amino acid side chain parameters for correlation studies in biology and pharmacology. Int J Pept Protein Res 32:269–278
Feng ZP (2002) An overview on predicting the subcellular location of a protein. In Silico Biol 2:291–303
Gao QB, Wang ZZ, Yan C, Du YH (2005a) Prediction of protein subcellular location using a combined feature of sequence. FEBS Lett 579:3444–3448
Gao Y, Shao SH, Xiao X, Ding YS, Huang YS, Huang ZD, Chou KC (2005b) Using pseudo amino acid composition to predict protein subcellular location: approached with Lyapunov index, Bessel function, and Chebyshev filter. Amino Acids 28:373–376
Gao WN, Wei DQ, Li Y, Gao H, Xu WR, Li AX, Chou KC (2007) Agaritine and its derivatives are potential inhibitors against HIV proteases. Med Chem 3:221–226
Gonzalez-Diaz H, Sanchez-Gonzalez A, Gonzalez-Diaz Y (2006) 3D-QSAR study for DNA cleavage proteins with a potential anti-tumor ATCUN-like motif. J Inorg Biochem 100:1290–1297
Gonzalez-Díaz H, Gonzalez-Díaz Y, Santana L, Ubeira FM, Uriarte E (2008) Proteomics, networks, and connectivity indices. Proteomics 8:750–778
Hong B, Tang QY, Yang FS (1999) Apen and Cross-ApEn: property, fast algorithm and preliminary application to the study of EEG and cognition. Signal Process 15:100–108 (in Chinese)
Hopp TP, Woods KR (1981) Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci USA 78:3824–3828
Huang Y, Li YD (2004) Prediction of protein subcellular locations using fuzzy k-NN method. Bioinformatics 20(1):21–28
Huang J, Shi F (2005) Support vector machines for predicting apoptosis proteins types. Acta Biotheor 53:39–47
Huang WL, Chen HM, Hwang SF, Ho SY (2006) Accurate prediction of enzyme subfamily class using an adaptive fuzzy K-nearest neighbor method. BioSystems 90(2):405–413. doi:10.1016/j.biosystems.2006.10.004
Janin J (1979) Surface and inside volumes in globular proteins. Nature 277:491–492
Janin J, Wodak S (1978) Conformation of amino acid side-chains in proteins. J Mol Biol 125:357–386
Kawashima S, Ogata H, Kanehisa M (1999) AAindex: amino acid index database. Nucleic Acids Res 27:368–369
Kedarisetti KD, Kurgan LA, Dick S (2006) Classifier ensembles for protein structural class prediction with varying homology. Biochem Biophys Res Commun 348:981–988
Keller JM, Gray MR, Givens JA (1985) A fuzzy k-nearest neighbors algorithm. IEEE Trans Syst Man Cybern 15:580–585
Kennedy J, Eberhart RC (1995) Particle swarm optimization. In: Proceedings of the 1995 IEEE International Conference on Neural Networks, vol 4, Perth, Australia, pp 1942–1948
Kennedy J, Eberhart RC (1997) A discrete binary version of the particles warm algorithm. Systems, man and cybernetics, 1997. In: Proceedings of the IEEE International Conference on Computational Cybernetics and Simulation, vol 5, October 12–15, pp 4104–4108
Kennedy J, Eberhart RC, Shi Y (2001) Swarm intelligence. Morgan Kaufman, San Mateo
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26:239–257
Li TT, Chou KC (1976) The quantitative relations between diffusion-controlled reaction rate and characteristic parameters in enzyme-substrate reaction system: 1. Neutral substrate. Sci Sinica 19:117–136
Li Y, Wei DQ, Gao WN, Gao H, Liu BN, Huang CJ, Xu WR, Liu DK, Chen HF, Chou KC (2007) Computational approach to drug design for oxazolidinones as antibacterial agents. Med Chem 3:576–582
Lin H, Li QZ (2007a) Predicting conotoxin superfamily and family by using pseudo amino acid composition and modified Mahalanobis discriminant. Biochem Biophys Res Commun 354:548–551
Lin H, Li QZ (2007b) Using pseudo amino acid composition to predict protein structural class: approached by incorporating 400 dipeptide components. J Comput Chem 28:1463–1466
Liu H, Wang M, Chou KC (2005a) Low-frequency Fourier spectrum for predicting membrane protein types. Biochem Biophys Res Commun 336:737–739
Liu H, Yang J, Wang M, Xue L, Chou KC (2005b) Using Fourier spectrum analysis and pseudo amino acid composition for prediction of membrane protein types. Protein J 24:385–389
Nakashima H, Nishikawa K (1994) Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J Mol Biol 238(1):54–61
Park KJ, Kanehisa M (2003) Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 19(13):1656–1663
Peter ME, Heufelder AE, Hengartner MO (1997) Advances in apoptosis research. Proc Natl Acad Sci USA 94:12736–12737
Pincus SM (1991) Approximate entropy as a measure of system complexity. Proc Natl Acad Sci USA 88:2297–2301
Prado-Prado FJ, Gonzalez-Diaz H, de la Vega OM, Ubeira FM, Chou KC (2008) Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for Input-Coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorg Med Chem 16:5871–5880
Reed JC, Paternostro G (1999) Postmitochondrial regulation of apoptosis during heart failure. Proc Natl Acad Sci USA 96:7614–7616
Richman JS, Moorman JR (2000) Physiological time-series analysis using approximate entropy and sample entropy. Am J Physiol Heart Circ Physiol 278(6):H2039–H2049
Schulz JB, Weller M, Moskowitz MA (1999) Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol 45:421–429
Shen HB, Chou KC (2006a) Ensemble classifier for protein fold pattern recognition. Bioinformatics 22:1717–1722
Shen HB, Chou KC (2006b) Using ensemble classifier to identify membrane protein types. Amino Acids 32(4):483–488. doi:10.1007/s00726-006-0439-2
Shen HB, Chou KC (2007a) Hum-mPLoc: an ensemble classifier for large-scale human protein subcellular location prediction by incorporating samples with multiple sites. Biochem Biophys Res Commun 355(4):1006–1011
Shen HB, Chou KC (2007b) EzyPred: a top-down approach for predicting enzyme functional classes and subclasses. Biochem Biophys Res Comm 364:53–59
Shen HB, Chou KC (2007c) Signal-3L: a 3-layer approach for predicting signal peptide. Biochem Biophys Res Comm 363:297–303
Shen HB, Chou KC (2008) HIVcleave: a web-server for predicting HIV protease cleavage sites in proteins. Anal Biochem 375:388–390
Shen HB, Yang J, Liu XJ, Chou KC (2005) Using supervised fuzzy clustering to predict protein structural classes. Biochem Biophys Res Commun 334:577–581
Shen HB, Yang J, Chou KC (2006) Fuzzy KNN for predicting membrane protein types from pseudo-amino acid composition. J Theor Biol 240:9–13
Shen HB, Yang J, Chou KC (2007a) Euk-PLoc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids 33(1):57–59
Shen HB, Yang J, Chou KC (2007b) Methodology development for predicting subcellular location and other attributes of proteins. Expert Rev Proteomics 4(4):453–463
Shi JY, Zhang SW, Pan Q, Cheng YM, Xie J (2007) Prediction of protein subcellular localization by support vector machines using multi-scale energy and pseudo amino acid composition. Amino Acids 33:69–74
Shi JY, Zhang SW, Pan Q, Zhou GP (2008) Using pseudo amino acid composition to predict protein subcellular location: approached with amino acid composition distribution. Amino Acids 35:321–327
Sirois S, Wei DQ, Du QS, Chou KC (2004) Virtual screening for SARS-CoV protease based on KZ7088 pharmacophore points. J Chem Inf Comput Sci 44:1111–1122
Steller H (1995) Mechanisms and genes of cellular suicide. Science 267:1445–1449
Tanford C (1962) Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J Am Chem Soc 84:4240–4274
Wang JF, Wei DQ, Chen C, Li Y, Chou KC (2008) Molecular modeling of two CYP2C19 SNPs and its implications for personalized drug design. Protein Pept Lett 15:27–32
Xiao X, Shao SH, Ding YS, Huang ZD, Huang YS, Chou KC (2005) Using complexity measure factor to predict protein subcellular location. Amino Acids 28:57–61
Xiao X, Shao SH, Ding YS, Huang ZD, Chou KC (2006a) Using cellular automata images and pseudo amino acid composition to predict protein subcellular location. Amino Acids 30(1):49–54
Xiao X, Shao SH, Huang ZD, Chou KC (2006b) Using pseudo amino acid composition to predict protein structural classes: approached with complexity measure factor. J Comput Chem 27:478–482
Zhang R, Wei DQ, Du QS, Chou KC (2006a) Molecular modeling studies of peptide drug candidates against SARS. Med Chem 2:309–314
Zhang TL, Ding YS, Chou KC (2006b) Prediction of protein subcellular location using hydrophobic patterns of amino acid sequence. Comput Biol Chem 30:367–371
Zhang ZH, Wang ZH, Zhang ZR, Wang YX (2006c) A novel method for apoptosis protein subcellular localization prediction combining encoding based on grouped weight and support vector machine. FEBS Lett 580:6169–6174
Zhang TL, Ding YS, Chou KC (2008) Prediction protein structural classes with pseudo amino acid composition: approximate entropy and hydrophobicity pattern. J Theor Biol 250(1):186–193
Zheng H, Wei DQ, Zhang R, Wang C, Wei H, Chou KC (2007) Screening for new agonists against Alzheimer’s Disease. Med Chem 3:488–493
Zhou GP (1998) An Intriguing controversy over protein structural class prediction. J Protein Chem 17:729–738
Zhou GP, Assa-Munt N (2001) Some insights into protein structural class prediction. Proteins 44:57–59
Zhou GP, Cai YD (2006) Predicting protease types by hybridizing gene ontology and pseudo amino acid composition. Proteins 63(3):681–684
Zhou GP, Doctor K (2003) Subcellular location prediction of apoptosis proteins. Proteins: Struct Funct Genet 50:44–48
Zhou XB, Chen C, Li ZC, Zou XY (2008) Improved prediction of subcellular location for apoptosis proteins by the dual-layer support vector machine. Amino Acids 35(2):383–388
Zimmerman JM, Eliezer N, Simha R (1968) The characterization of amino acid sequences in proteins by statistical methods. J Theor Biol 21:170–201
Acknowledgments
The authors wish to thank Dr. Z. H. Zhang for providing the datasets. This work was supported in part by Specialized Research Fund for the Doctoral Program of Higher Education from Ministry of Education of China (No. 20060255006), Project of the Shanghai Committee of Science and Technology (No. 08JC1400100), Shanghai Talent Developing Foundation (No. 001), Specialized Foundation for Excellent Talent from Shanghai, and the Open Fund from the Key Laboratory of MICCAI of Shanghai (06dz22013).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gu, Q., Ding, YS., Jiang, XY. et al. Prediction of subcellular location apoptosis proteins with ensemble classifier and feature selection. Amino Acids 38, 975–983 (2010). https://doi.org/10.1007/s00726-008-0209-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-008-0209-4