
Abstract
A nitrogen-related signal transduction pathway, consisting of the three phosphotransfer proteins EINtr, NPr, and IIANtr, was discovered recently to regulate the uptake of K+ in Escherichia coli. In particular, dephosphorylated IIANtr inhibits the activity of the K+ transporter TrkA. Since the phosphorylation state of IIANtr is partially determined by its reversible phosphorylation by NPr, we have determined the three-dimensional structure of NPr by solution NMR spectroscopy. In total, we obtained 973 NOE-derived distance restraints, 112 chemical shift-derived backbone angle restraints, and 35 hydrogen-bond restraints derived from temperature coefficients (wave). We propose that temperature wave is useful for identifying exposed beta-strands and assists in establishing protein folds based on chemical shifts. The deduced structure of NPr contains three α-helices and four β-strands with the three helices all packed on the same face of the β-sheet. The active site residue His16 of NPr for phosphoryl transfer was found to be neutral and in the Nε2-H tautomeric state. There appears to be increased motion in the active site region of NPr compared to HPr, a homologous protein involved in the uptake and regulation of carbohydrate utilization.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Potassium is the major intracellular cation in all living cells. In bacteria, cytoplasmic K+ is required in a variety of biological processes by serving as (1) an osmotic solute, (2) an activator of intracellular enzymes, (3) a regulator of internal pH, and (4) a second messenger (Epstein 2003). In standard media, E. coli maintains a cytoplasmic K+ concentration in the range of 300–500 mM. Of note, there are only a few types of transporters specific for K+. Depending on the level of K+ available in the environment, bacteria utilize different transporters. When the concentration of K+ is low, K+ uptake is achieved via a high-affinity ATPase (the Kdp transporter, K m ∼ 2 μM). Under normal conditions, Kdp is inhibited and two constitutive transporters, Trk and Kup, are used for K+ uptake. The K+ affinities (K m) of Trk and Kup are both ∼1 mM. However, Trk is a faster system than Kup with a V max for K+ uptake of 200–500 μmol min−1 g−1. The Trk system consists of several proteins: TrkA, TrkE, and TrkG. While TrkE and TrkG are integral membrane proteins, TrkA is a peripheral protein that binds to the inner membrane surface with the aid of other proteins (Fig. 1). Most point mutations of TrkA disrupted its K+ transport activity, indicating that this protein is essential for K+ uptake (Bossemeyer et al. 1989; reviewed by Epstein 2003).

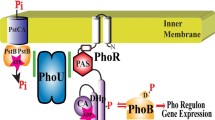
The regulation of bacterial K+ uptake by a protein-mediated signal transduciton pathway. In this protein pathway, the phosphoryl group provided by a high-energy molecule phosphoenolpyruvate (PEP) is transferred from nitrogen-related enzyme I (EINtr) to NPr and then from NPr to IIANtr. The GAF domain in EINtr is unique in the nitrogen PTS pathway and may serve as a sensor for nitrogen-containing compounds yet to be identified (Rabus et al. 1999; Peterkofsky et al. 2006). Phosphorylated IIANtr dissociates from the bacterial potassium transporter TrkA, allowing the uptake of K+ under normal conditions. When IIANtr is dephosphorylated, however, the protein blocks K+ uptake (Lee et al. 2007)
Sequence analysis revealed that TrkA is composed of two similar halves. The N-terminal region of each half (residues 1–130 and 234–355) is similar to the complete NAD-binding domain of NAD-dependent dehydrogenases. The C-terminal region of each half (residues 131–233 and 357–458) shows sequence similarity to the first 100 residues of the catalytic domain of glyceraldehyde-3-phosphate dehydrogenase. Based on these results, a model was built for TrkA (Schlosser et al. 1993). A glycine-rich motif around residues 240–250 is proposed as the NAD binding site (Bellamacina 1996). Recently, Jiang et al. (2001) determined the structure of the intracellular C-terminal domain of the E. coli K+ channel by X-ray diffraction and found a unique Rossmann-fold with a well-conserved salt-bridge and a hydrophobic dimer interface. This structure represents a broad class of domains/proteins important for regulating conductance of K+ (referred to as RCK domain) in both prokaryotic K+ transporters and K+ channels (collectively termed the transportome) (MacKinnon 2003; Gouaux and MacKinnon 2005). Remarkably, RCK domains are also found in a subfamily of eukaryotic K+ channels, the large-conductance Ca2+-activated K+ channels. In particular, Jiang et al. (2001) also found that the sequence of the C-terminal half of the E. coli K+ transport protein TrkA aligns with the RCK domain of the E. coli K+ channel. Such information provides an excellent basis for computational modeling of membrane protein structure (see Fig. 1) as well as structure-based drug design to combat pathogenic bacteria such as E. coli (Chou 2004a, 2004b). The open and closed states of bacterial transporters or channels are highly regulated and critically important in potassium homeostasis. In the case of the E. coli transport protein TrkA, however, which protein regulates its activity was not elucidated until 2007.
A bacterial nitrogen regulatory pathway (Fig. 1), consisting of the three protein members EINtr, NPr and IIANtr, was described in the 1990s (Powell et al. 1995; Rabus et al. 1999). Of note, these proteins were found to show sequence homology to the classical phosphoenolpyruvate:sugar phosphotransferase system (PTS) (reviewed by Peterkofsky et al. 2006). This classical PTS pathway contributed significantly to our understanding of protein-mediated signal transduciton via phosphorylation and dephosphorylation. Indeed, phosphorylation and dephosphrylation of proteins are of general importance in regulating a variety of physiological processes (e.g., see Surks et al. 1999). Of particular interest is that NPr and IIANtr are co-transcribed with σ54 (Powell et al. 1995) and negatively regulate this sigma factor, important for transcription of genes for nitrogen metabolism (Merrick and Coppard 1989); this finding led to the suggestion that the pathway plays a role in nitrogen regulation. IIANtr homologues have been identified in several other Gram-negative bacteria.
Recently, it was found that E. coli harboring a deletion of ptsN (encoding IIANtr) became extremely sensitive to leucine-containing peptides (LCP), while strains deleted for ptsO (encoding EINtr) or ptsP (encoding NPr) were more resistant than the wild type. The toxicity due to leucine accumulation could be neutralized by dephosphorylated IIANtr (Lee et al. 2005). This form of IIANtr was shown to be required for derepression of E. coli K12 ilvBN expression. The ilvBN operon encodes acetohydroxy acid synthase (AHAS) I, which catalyzes the first step common to the biosynthesis of the branched-chain amino acids. This discovery underscores the importance of the phosphorylation state of the nitrogen pathway in regulating amino acid synthesis and bacterial growth. Subsequently, IIANtr was found to associate tightly with the K+ transport protein TrkA and only dephosphorylated IIANtr inhibited the activity of TrkA (Lee et al. 2007). The discovery of Lee et al., therefore, established a critical biological link between the previously described nitrogen PTS pathway and the K+ transporter in E. coli as summarized in Fig. 1.
To provide insight into the mechanism of phosphoryl transfer in the nitrogen pathway and its regulation of bacterial K+ uptake, one important approach is to determine the structures of the relevant proteins and their complexes, including membrane proteins (Fig. 1), many of which are key targets for drug discovery (Peterkofsky et al. 2001; MacKinnon 2003; Schnell and Chou 2008; Wang 2008). To date, only the structure of IIANtr, determined by crystallography, in this bacterial signaling pathway was reported (Bordo et al. 1998). Using residual dipolar coupling and chemical shift data, our previous NMR studies revealed that this protein is monomeric in solution and only molecule A in the dimeric crystal structure resembles the solution structure (Li et al. 2003; Wang et al. 2005). Here, we report the cloning, expression, purification, and structural determination of NPr, a key member of the nitrogen pathway (Fig. 1) that directly communicates with and controls the phosphorylation state of IIANtr.
Materials and methods
Cloning, expression and purification of NPr
NPr is a protein of 90 residues. The coding sequence was first cloned into the expression vector pRE1 (Reddy et al. 1989). E. coli K12 chromosomal DNA was used as the template for a PCR reaction with the forward primer 5′-TGGAAAAACGTAACAT ATGACCGTCAAGCAAACTGTTGAAA-3′ (NdeI site underlined, the first codon of coding sequence in bold) and the reverse primer, approximately 250 bp downstream of the NPr termination codon, 5′-CATAACATTTCCGATGTCTAGAGGTGCCCAGGGGACTTTG-3′ (XbaI site underlined). After restriction with the appropriate enzymes, the NdeI-XbaI fragment was cloned into pRE1 and transformed into GI698 (LaVallie et al. 1993). The recovery of expressed protein in this clone was very poor so alternative strategies were explored. As described previously (Wang et al. 2005), successful expression was achieved by an intein expressed protein, containing an additional C-terminal five residues from cloning; we reported earlier that this C-terminal extension was disordered. Further experiments indicated that the last five residues of the native structure showed few NOE cross peaks (G.W. and A.P., unpublished results). Consequently, a truncated version of NPr (residues 1–85) was engineered. A reverse primer (5′-GAAGATTAATCTAGATCAAATCAAGAATTAAAGAGG-3′) encoding an XbaI cloning site (underlined) and a stop codon (italics) immediately after codon 85 was used together with a forward primer upstream of the translation start site in a PCR reaction with the pRE1-NPr clone as the template. The PCR product was cloned by standard procedures into the NdeI and AvrII sites of pET-Duet-1 (Novagen), and the recombinant vector was transformed into a derivative of strain ER2566 (New England Biolabs) in which the pts operon was inactivated by a kanamycin gene insert. The recombinant plasmid was verified by DNA sequencing.
A 2-l culture of ER2566(Δpts)/pET-Duet-1-NPr(1–85) in LB/Amp/Kan was induced with IPTG at A 600 = 0.4 and allowed to grow overnight at 30°C. The washed pellets were extracted with Bug Buster Master Mix (Novagen) and the supernatant solutions were fractionated on a MonoQ 10/10 column (10 mM Tris, pH 7.5/0.5 mM EDTA; gradient = 0–500 mM NaCl). The fractions enriched in NPr were pooled and concentrated (Filtron 3K, Pall, East Hills, N.Y.), then further purified on a Superdex column (120-ml bed volume; buffer = 10 mM Tris, pH 7.5/0.5 mM EDTA). The resultant protein was essentially pure, as judged by SDS-PAGE stained with Coomassie blue. The yield of the purified protein was approximately 50 mg. A stock solution of purified NPr (2.5 mg/ml) was stored at −80°C until use. The isolated protein was demonstrated to be active as a phosphoryl acceptor from EINtr and was also capable of phosphotransfer to IIANtr. The stock solution was further concentrated to obtain an NMR sample containing ∼1 mM protein, 25 mM Tris buffer at pH ∼ 7.
NMR spectroscopy
All data were collected at 35°C on a three-channel Varian INOVA 600-MHz NMR instrument with waveform generators and triple-axis pulsed field gradient accessories. Several double- and triple-resonance NMR experiments were performed, including HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, C(CO)NH, H(CCO)NH, HNHA, and HBHA(CO)NH (Bax and Grzesiek 1993; Kay 1997). HCCH-COSY, HCCH-TOCSY and 3D 15N- or 13C-separated NOESY spectra were also collected. To verify the NOE assignments, a 4D 15N- and 13C-edited NOESY spectrum was also recorded. A DIPSI-3 isotropic mixing time of 12 ms was used for the HN-detected TOCSY unit and 100 ms was employed in the NOESY experiment. Typically, 3D experiments were recorded with sweep widths (increments) of 2,200 Hz (28) for 15N, 12,067.8 Hz (60) for aliphatic 13C, and 3,770 Hz (40) for carbonyl carbon in the indirect dimensions, and 8,510.6 and 1,024 complex points in the 1H-detected dimension, respectively. In the case of an indirect proton dimension, 90–120 increments were collected. The carriers for 1H, 15N, and 13C were positioned at 4.67, 118.2, and 47.3 ppm, respectively. Data were processed using NMRPipe (Delaglio et al. 1995) and analyzed by PIPP (Garrett et al. 1991). 1H chemical shifts were referenced to DSS and 13C and 15N chemical shifts were referenced according to the IUPAC recommendation (Markley et al. 1998).
In all 2D NMR experiments such as HSQC, the sweep width for the 15N dimension was typically 2,200 Hz with 100 increments, whereas 1,024 complex points were collected in the 1H-detected dimension with a spectral width of 8,510.6 Hz. The chemical shifts for amide protons were measured by two-dimensional HSQC every 5° from 10 to 35°C. The cross peaks were picked based on the assignments achieved above using PIPP. The temperature coefficient for each backbone amide proton of the protein was calculated by linear regression of chemical shifts versus temperature and represented in ppm × 10−3/°C (or ppt/°C).
Residue-specific heteronuclear 15N{1H} NOE values for NPr and HPr were measured as described previously (Wang et al. 2005) from 2D (1H, 15N) correlated spectroscopy with and without proton saturation. Heteronuclear NOE values were obtained by taking the ratios of the peak intensities in the above two experiments (Kay et al. 1989; Wang et al. 1999).
Structural determination
Previous studies (Anderson et al. 1991; Kruse et al. 1993) have indicated the importance of C-terminal residues in the HPr from E. coli but not from S. carnosus. We demonstrated (A. Peterkofsky and R.L. Levine, unpublished studies) that NPr85 is active both in phosphoryl acceptance from EINtr as well as in phosphotransfer to IIANtr. Thus, we concluded that the truncated form of NPr was relevant for structural and functional studies. The solution structure of NPr was determined using the established approach (Wüthirch 1986). The major structural restraints were derived from different NOESY spectra. These include 3D 15N-separated NOESY, 13C-separated NOESY, and 4D 15N- and 13C-edited NOESY spectra. The cross peaks were integrated by PIPP (Garrett et al. 1991) and converted to distance restraints (1.8–2.8, 1.8–3.8, 1.8–5.0, and 1.8–6.0 Å corresponding to strong, medium, weak, and very weak types of NOE peaks) (Clore and Gronenborn 1998). Backbone angles were obtained from TALOS analysis of sets of backbone heteronuclear chemical shifts, including 1H, 15N, 13Cα, 13Cβ, and 13CO (Cornilescu et al. 1999). A broader range (±20° or greater) than predicted was allowed for each angle in the structural calculations. Hydrogen bond restraints for the structured regions were derived from temperature coefficients using the criteria (>−4.5 ppt/°C) (Baxter and Williamson 1997; Cierpicki and Otlewski 2001). Each hydrogen bond was converted to two distance restraints. An extended covalent structure was used as starting coordinates. The initial protein fold was established based on unambiguous NOE connectivities between the chemical shift-derived secondary structures. This structure was then used as an aid for the assignments of additional NOE cross peaks to improve the quality of the structure (Wang et al. 1999). An ensemble of structures was calculated by using the simulated annealing protocol in the Xplor-NIH program (Schwieters et al. 2003). The structures accepted have no NOE violations greater than 0.50 Å, rmsd for bond deviations from ideality less than 0.01 Å, and rmsd for angle deviations from ideality less than 5°. The structures were viewed and analyzed using MOLMOL (Koradi et al. 1996) and PROCHECK (Laskowski et al. 1993).
Results
NPr constructs and the tautomeric state of the active-site histidine
Initially, NPr was expressed as a fusion protein using the intein technology and the final construct contained five additional residues at the C-terminus (referred to as NPr95), since the overexpression of unmodified NPr was problematic (Wang et al. 2005). NMR analysis found that the five additional residues at the C-terminus of NPr95 from molecular cloning are disordered in solution (Wang et al. 2005). Furthermore, some residues of this form of NPr showed two sets of NMR peaks at elevated concentrations in HSQC spectra, probably due to the influence of the C-terminal disordered tail. Two sets of peaks were also detected for NPr95 in a simpler HSQC spectrum that gave 1H–15N correlated peaks only for histidine rings (Fig. 2a). NPr contains only one histidine and the two sets of peaks indicate two forms of His16 in the protein construct. To improve the spectral quality, we also made a truncated form of NPr by deleting the C-terminal five residues. This NPr construct contains 85 residues and is referred to hereinafter as NPr85. Satisfyingly, NPr85 gave only one set of cross peaks (Fig. 2b). Furthermore, the HSQC patterns of NPr85 and NPr95 are very similar (not shown), indicating a similar folded structure. Previously, we reported that NPr95 is capable of interacting with IIANtr (Wang et al. 2005). Our ongoing NMR studies (unpublished) reveal that NPr85 can bind either IIANtr or EINtr with only small chemical shift changes. These results indicate that NPr85 has a relevant structure and is competent in interacting with its partner proteins. The protein is also active in both phosphoacceptance and phosphotransfer (see “Materials and methods”). As a consequence, NPr85 was used for all subsequent NMR analysis.

Long-range 1H and 15N correlations for the His16 ring of a NPr95 and b NPr85. Note that a second set of cross peaks (connected with dotted lines and labeled with H16′) was detected in NPr95 but not in NPr85. See the text for further details
The protonation or deprotonation of the histidine ring influences its chemical shifts. A neutral form has a nitrogen chemical shift at ∼168 ppm, while the unprotonated form appears at ∼250 ppm. In contrast, both nitrogen nuclei in a positively charged histidine have similar chemical shifts at ∼175 ppm with a ∼1-ppm separation between them (Pelton et al. 1993). In the case of the only histidine of NPr, the chemical shift separation between the two imidazole nitrogens is ∼40 ppm, indicating that His16 is neutral. Furthermore, the peak pattern shown in Fig. 2b indicates the presence of the Nε2-H tautomeric state. The Nε2-H tautomeric state of a histidine was found to be more stable than the Nδ1-H form (Pelton et al. 1993; Wang et al. 2005).
1H, 15N, and 13C chemical shift assignments of NPr85 and secondary structures
Chemical shifts of NPr85 were assigned using a procedure described elsewhere (Li et al. 2003). In brief, backbone chemical shifts of NPr85 were assigned using a suite of triple-resonance experiments (see “Materials and methods”). Side chain signals were assigned based on HBHA(CO)NH, C(CO)NH, H(CCO)NH, HCCH-COSY, and HCCH-TOCSY experiments.
To elucidate the potential α-helices and β-strands along the polypeptide chain, Hα secondary shifts were calculated and plotted against residue number in Fig. 3. Secondary shifts (ΔHα) are the chemical shift differences between measured and random-coil shifts (Wishart and Sykes 1994). The random coil values were taken from Wüthrich (1986). A group of negative bars (upfield shifts <−0.1 ppm) for ΔHα suggests helical structures and a train of positive bars (>0.1 ppm) suggests β-strands. Thus, the chemical-shift-derived helical regions in NPr85 are located at residues 17–26 (α1), 49–56 (α2), and 72–83 (α3), while the β-strands are found for residues 2–8 (β1), 32–38 (β2), 43–45 (β3), and 61–69 (β4) (Fig. 3).

The Hα secondary shifts of NPr85 at pH 7.0 and 35°C. See the text for the definition of secondary shifts. The cutoff lines for the helical and β-stranded regions are placed at −0.1 and +0.1 ppm, respectively (Wishart et al. 1991). The chemical shift-derived three α-helical and four β-stranded regions of NPr85 are indicated
Three-dimensional structure of NPr85
The structure of NPr85 was determined based on the following NMR restraints: NOE-derived distance restraints, chemical shift derived backbone angle restraints, and hydrogen-bond restraints (see “Materials and methods”). As listed in Table 1, there are 973 distance restraints, 112 backbone angle restraints, and 35 hydrogen bond restraints. Hydrogen bonded amide protons were identified from temperature wave (a plot of temperature coefficients versus protein residue number) (Fig. 4) only for the residues in the structured regions (helices and strands). In total, 50 structures were calculated and 30 of them that passed the structural acceptance criteria (see “Materials and methods”) were subject to further analysis. The rmsd was 0.53 Å when the backbone atoms of the entire protein chain (residues 1–85) were superimposed. When the rmsd value for each of the structured regions (i.e., 3 helices and 4 strands) was measured, β-strands (0.058–0.11 Å) were found to be slightly better defined than helices (0.1–0.25 Å). Few NOE restraints were observed for the region before α1 (Fig. 5a), where the active-site histidine is located, leading to a poorly defined region. According to PROCHECK analysis, the backbone angles of 97.7% of the residues are located in the allowed region. From the ribbon diagram in Fig. 5b, it is clear that the helical regions are located between residues 18–27 (α1), 49–56 (α2), and 72–83 (α3), whereas the β-strands are located between residues 2–8 (β1), 35–38 (β2), 42–45 (β3), and 62–67 (β4). Hence, these calculated structural regions agree well with those derived from secondary chemical shifts in Fig. 3. In the 3D structure, the three helices are all packed on the same side of the β-sheet consisting of the four β-strands.

Temperature wave of the backbone amide protons of NPr85. Temperature wave is defined as the change of temperature coefficients of the backbone amide protons of a protein as a function of residue number. Missing values are due to either fast exchange or prolines (P19 and P70). Values greater than −4.5 ppt/°C are regarded as hydrogen bonded (Baxter and Williamson 1997; Cierpicki and Otlewski 2001). We noted that the wave patterns for exposed and buried strands differ (see “Discussion”)

Three-dimensional structure of NPr85 determined by solution NMR spectroscopy. See Table 1 for the statistical data of the structure. a The backbone atoms of residues 1–85 of an ensemble of 30 structures (out of 50 calculated) were superimposed to illustrate the precision of the structure. The N-, C-termini, and the active site residue His16 are labeled. b Ribbon diagram of a representative structure of NPr85 indicates the three helices and four β-strands
Heteronuclear 15N NOE measurements
To shed additional light on the structure and function of the protein, we also measured heteronuclear nuclear Overhauser effects (NOEs) for NPr85 (Fig. 6a). As the majority of heteronuclear NOE values are greater than 0.6, the protein chain is folded with backbone motions on the ps-ns timescale (Kay et al. 1989; Wang et al. 2005). Two residues at the C-terminus of this protein construct have NOE values less than 0.6, suggesting increased motion. Another region with an NOE value less than 0.6 (Fig. 6a) coincides with the poorly defined active-site region (Fig. 5a), indicating a correlation between motion and poor structural precision in this case. Furthermore, due to line broadening, the intensities of the cross peaks in HSQC spectra for those residues are weaker than those in other regions, indicative of motions on the ms–μs time scale. Such motions have been shown to be essential for protein–protein interactions (Kay et al. 1989; Peterkofsky et al. 2001; Marintchev et al. 2007; Hansen et al. 2008) as observed previously for IIANtr in the presence of NPr (Wang et al. 2005).

15N{1H} NOE values of a NPr85 and b HPr measured using 15N-labeled proteins at pH 7 and 35°C on the Varian INOVA 600-MHz NMR spectrometer equipped with a cryogenic probe
Comparison with HPr, a phosphoryl transfer protein involved in glucose utilization
NPr was initially identified by bioinformatics based on sequence homology to HPr, a protein of similar size involved in carbohydrate utilization (Peterkofsky et al. 2006). The sequence identity between the two proteins is ∼27%. Our structural determination made it clear that NPr85 indeed adopts a protein fold similar to HPr (Klevit and Waygood 1986; Jia et al. 1993). To provide additional insight into the differences between the two proteins, we also measured 15N NOE values for HPr (Fig. 6b). The heteronuclear NOE values in the HPr case (on average 0.76) are slightly more positive than those of NPr85 (on average 0.72) (Fig. 6a). In other words, NPr85 is slightly more mobile than HPr, especially in the region near the active site and the C-terminus of the protein as indicated by an NOE value less than 0.6. The more pronounced line broadening of the peaks for NPr85 residues in the vicinity of the active site indicates additional motion on the ms–μs time scale.
Discussion
We have made several NPr constructs, and NPr85 was active as both a phosphoacceptor and phosphodonor and found to possess favorable properties for NMR analysis. Structural determination of NPr85 revealed an αβ-fold with three helices packed onto a four-stranded β-sheet (Fig. 5B). In the 3D structure, β1 and β3 are exposed, while β2 and β4 are located in the interior of the sheet. Correspondingly, nearly all of the temperature coefficients in the β2 and β4 regions (except for residue 62) are above the −4.5 ppt/°C cutoff line (Fig. 4) as a result of hydrogen bonding with two neighboring strands. For β1 and β3, the temperature coefficients are distributed above and below the cutoff line, consistent with an alternative pattern of hydrogen bonding in these two strands (i.e., with water and with the adjacent strand). We are continuing to investigate the generality of this observation; such a temperature wave pattern (Fig. 4) may be useful in identifying solvent exposed β-strands as an aid in establishing spatial relationships between strands identified from chemical shift analysis (Fig. 3).
The 3D structure of NPr provides a basis for us to understand the specific interactions of this protein with its upstream and downstream protein partners in the nitrogen pathway that regulates bacterial K+ transport (Fig. 1). In addition to structure, NMR is capable of providing other unique information. Determination of the chemical form of the active site histidine is an important prerequisite for elucidating the mechanism of the protein-mediated phosphoryl transfer process (Wang et al. 2000; Peterkofsky et al. 2001). The active site residue His16 of NPr was found to be in the Nε2 tautomeric state (Fig. 2).
NPr was found to adopt a protein fold similar to that of HPr from the carbohydrate pathway. Importantly, Rabus et al. (1999) found that HPr did not interact with EINtr from the nitrogen pathway, and NPr from the nitrogen pathway did not interact with enzyme I (EI) from the carbohydrate pathway. Our NMR study also provided information concerning protein dynamics. Such information, which is not readily available from X-ray diffraction, is essential in understanding protein function as well as protein regulation of various biological processes (Chou et al. 2001; Zhou and Troy 2005). Because NPr85 is slightly more mobile than HPr (Fig. 6), we propose that such differences, along with the sequence differences between HPr and NPr, may be important in avoiding crosstalk between the two signal transduction pathways.
Abbreviations
- NPr:
-
Nitrogen-related HPr
- HPr:
-
Histidine-containing protein
- EINtr :
-
Nitrogen-related enzyme I
- HSQC:
-
Heteronuclear single quantum coherence spectroscopy
- IIANtr :
-
Nitrogen-related enzyme IIA
- GAF:
-
cGMP-specific and –stimulated phosphodiesterases, Anabaena adenylate cyclases and Escherichia coli FhlA
- IPTG:
-
Isopropyl-β-D-thiogalactopyranoside
- NMR:
-
Nuclear magnetic resonance
- NOE:
-
Nuclear Overhauser effect
- PCR:
-
Polymerase chain reaction
- PEP:
-
Phosphoenolpyruvate
- PTS:
-
Phosphoenolpyruvate:sugar phosphotransferase system
- RCK:
-
Regulating conductance of K+
- rmsd:
-
Root mean square deviation
- SDS-PAGE:
-
Sodium dodecylsulfate-polyacrylamide gel electrophoresis
References
Anderson JW, Bhanot P, Georges F, Klevit RE, Waygood EB (1991) Involvement of the carboxy-terminal residues in the active site of the histidine-containing protein, HPr, of the phosphoenolpyruvate:sugar phosphotransferase system of Escherichia coli. Biochemistry 30:9601–9607
Bax A, Grzesiek S (1993) Methodological advances in protein NMR. Acc Chem Res 26:131–138
Baxter NJ, Williamson MP (1997) Temperature dependence of 1H chemical shifts in proteins. J Biomol NMR 9:359–369
Bellamacina CR (1996) The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins. FASEB J 10:1257–1269
Bordo D, van Monfort RLM, Pijning T et al (1998) The three-dimensional structure of the nitrogen regulatory protein IIANtr from Escherichia coli. J Mol Biol 279:245–255
Bossemeyer D, Borchard A, Dosch DC et al (1989) K+-transport protein TrkA of E. coli is a peripheral membrane protein that requires other trk gene products for attachment to the cytoplsmic membrane. J Biol Chem 264:16403–16410
Chou KC (2004a) Insights from modelling three-dimensional structures of the human potassium and sodium channels. J Proteome Res 3:856–861
Chou KC (2004b) Structural bioinformatics and its impact to biomedical science. Curr Med Chem 11:2105–2134
Chou JJ, Li S, Klee CB, Bax A (2001) Solution structure of Ca2+-calmodulin reveals flexible hand-like properties of its domains. Nat Struct Biol 8:990–997
Cierpicki T, Otlewski J (2001) Amide proton temperature coefficients as hydrogen bond indicators in proteins. J Biomol NMR 21:249–261
Clore GM, Gronenborn AM (1998) Determining structures of larger proteins and protein complexes. Trends Biotechnol 16:22–34
Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302
Delaglio F, Grzesiek S, Vuister GW et al (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Epstein W (2003) The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol 75:293–320
Garrett DS, Powers R, Gronenborn AM, Clore GM (1991) A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J Magn Reson 95:214–220
Gouaux E, MacKinnon R (2005) Principles of selective ion transport in channels and pumps. Science 310:1461–1465
Hansen DF, Vallurupalli P, Lundstrom P et al (2008) Probing chemical shifts of invisible states of proteins with relaxation dispersion NMR spectroscopy: how well can we do? J Am Chem Soc 130:2734–2735
Jia Z, Quail JW, Waygood EB, Delbaere LT (1993) The 2.0 Å resolution structure of the Escherichia coli histidine-containing phosphocarrier protein HPr: a redetermination. J Biol Chem 268:22940–22501
Jiang Y, Pico A, Cadene M et al (2001) Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in human BK channel. Neuron 29:593–601
Kay LE (1997) NMR methods for the study of protein structure and dynamics. Biochem Cell Biol 75:1–15
Kay LE, Torchia DA, Bax A (1989) Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry 28:8972–8979
Klevit RE, Waygood EB (1986) Two-dimensional 1H NMR studies of histidine-containing protein from Escherichia coli. 3. Secondary and tertiary structure as determined by NMR. Biochemistry 25:7774–7781
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14:51–55
Kruse R, Hengstenberg W, Beneicke W, Kalbitzer HR (1993) Involvement of various amino- and carboxyl-terminal residues in the active site of the histidine-containing protein HPr of the phosphoenolpyruvate-dependent phosphotransferase system of Staphylococcus carnosus: site-directed mutagenesis with the ptsH gene, biochemical characterization and NMR studies of the mutant proteins. Protein Eng 6:417–423
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structure. J Appl Cryst 26:283–291
LaVallie ER, DiBlasio EA, Kovacs S et al (1993) A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (NY) 11:187–193
Lee CR, Koo BM, Cho SH et al (2005) Requirement of the dephospho-form of enzyme IIANtr for derepression of Escherichia coli K-12 ilvBN expression. Mol Microbiol 58:334–44
Lee CR, Cho S-H, Yoon MJ, Peterkofsky A, Seok YJ (2007) Escherichia coli IIANtr regulates the K+ transporter TrkA. Proc Natl Acad Sci USA 104:4124–4129
Li X, Peterkofsky A, Wang G (2003) 1H, 15N, and 13C chemical shift assignments of the Escherichia coli nitrogen regulatory phosphocarrier IIANtr. J Biomol NMR 27:401–402
MacKinnon R (2003) Potassium channels. FEBS Lett 555:62–65
Marintchev A, Frueh D, Wagner G (2007) NMR methods for studying protein-protein interactions involved in translation initiation. Methods Enzymol 430:283–331
Markley JL, Bax A, Arata Y et al (1998) Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J Biomol NMR 12:1–23
Merrick MJ, Coppard JR (1989) Mutations in genes downstream of the rpoN gene (encoding σ54) of Klebsiella pneumoniae affect expression from σ54-dependent promoters. Mol Microbiol 3:1765–1775
Pelton JG, Torchia DA, Meadow ND, Roseman S (1993) Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci 2:543–558
Peterkofsky A, Wang G, Garrett DS et al (2001) Three-dimensional structures of protein–protein complexes in the E. coli PTS. J Mol Microbiol Biotechnol 3:347–354
Peterkofsky A, Wang G, Seok Y-J (2006) Parallel PTS systems. Arch Biochem Biophys 453:99–105
Powell BS, Court DL, Inada T et al (1995) Novel proteins of the phosphotransferase system encoded within the rpoN operon of Escherichia coli. Enzyme IIANtr affects growth on organic nitrogen and the conditional lethality of an erats mutant. J Biol Chem 270:4822–4839
Rabus R, Reizer J, Paulsen I, Saier MH Jr (1999) Enzyme INtr from Escherichia coli. A novel enzyme of the phosphoenolpyruvate-dependent phosphotransferase system exhibiting strict specificity for its phosphoryl acceptor, NPr. J Biol Chem 274:26185–26191
Reddy P, Peterkofsky A, McKenney K (1989) Hyperexpression and purification of Escherichia coli adenylate cyclase using a vector designed for expression of lethal gene products. Nucleic Acids Res 17:10473–10488
Schlosser A, Hamann A, Bossemeyer D et al (1993) NAD+ binding to the Escherichia coli K+-uptake protein TrkA and sequence similarity between TrkA and domains of a family of dehydrogenases suggest a role for NAD+ in bacterial transport. Mol Microbiol 9:533–543
Schnell JR, Chou JJ (2008) Structure and mechanism of the M2 proton channel of influenza A virus. Nature 451:591–595
Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160:65–73
Surks HK, Mochizuki N, Kasai Y et al (1999) Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Ialpha. Science 286:1583–1587
Wang G (2008) NMR of membrane-associated peptides and proteins. Curr Protein Pept Sci 9:50–69
Wang G, Wylie GP, Twigg PD et al (1999) Solution structure and peptide binding studies of the C-terminal Src homology 3-like domain of the diphtheria toxin repressor protein. Proc Natl Acad Sci USA 96:6119–6124
Wang G, Louis JM, Sondej M et al (2000) Solution structure of the phosphoryl transfer complex between the signal transducing proteins HPr and IIAglucose of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system. EMBO J 19:5635–5649
Wang G, Peterkofsky A, Keifer PA, Li X (2005) NMR characterization of the Escherichia coli nitrogen regulatory protein IIANtr in solution and interaction with its partner protein, NPr. Protein Sci 14:1082–1090
Wishart DS, Sykes BD (1994) The 13C chemical shift index: a simple method for the identification of protein secondary structure using 13C chemical shift data. J Biomol NMR 4:171–180
Wishart DS, Sykes BD, Richards FM (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol 222:311–333
Wüthrich K (1986) NMR of proteins and nucleic acids. Wiley, New York
Zhou GP, Troy FA (2005) NMR studies on how the binding complex of polyisoprenol recognition sequence peptides and polyisoprenols can modulate membrane structure. Curr Protein Pept Sci 6:399–411
Acknowledgments
This research was supported by the startup fund from the Eppley Institute of the University of Nebraska Medical Center (UNMC) to G.W. and, in part, by the Intramural Research Program of the NIH, NHLBI to A.P. The support of UNMC also includes the accessibility to the NMR Core Facility (supported by both a CORE grant from the National Cancer Institute-NIH and the Nebraska Research Initiative NRI). We are grateful to Frank Delaglio and Dan Garrett (NIH) for NMR software. We thank Paul Keifer for maintaining the NMR hardware during this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, X., Peterkofsky, A. & Wang, G. Solution structure of NPr, a bacterial signal-transducing protein that controls the phosphorylation state of the potassium transporter-regulating protein IIANtr . Amino Acids 35, 531–539 (2008). https://doi.org/10.1007/s00726-008-0079-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-008-0079-9