
Abstract
X-band electron spin relaxation times of BDPA (1:1 α,γ-bisdiphenylene-β-phenylallyl), galvinoxyl 2,6-di-tert-butyl-α-(3,5-di-tert-butyl-4-oxo-2,5-cyclohexadien-1-ylidene)-p-tolyloxy, DPPH (2,2-diphenyl-1-picrylhydrazyl) and thianthrene radicals in fluid solution were measured by electron spin echo and inversion recovery at ambient temperature. Tumbling correlation times are estimated to be in the range of 20–30 ps. In this fast tumbling regime T 1 ~ T 2. Relaxation times are compared with previously reported values for symmetrically substituted triarylmethyl, semiquinone, and nitroxide radicals. The concentration dependence of spin lattice relaxation for neutral BDPA in toluene is about 103 times greater than for anionic trityl radicals in water. T 1 decreases in the order carbon-center BDPA > galvinoxyl > DPPH > thianthrene. The dominant relaxation mechanisms are proposed to be a local mode for BDPA, spin rotation, local mode and modulation of anisotropic proton hyperfine coupling for galvinoxyl, modulation of anisotropic nitrogen hyperfine for DPPH, and spin rotation plus modulation of anisotropic proton hyperfine coupling for thianthrene.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
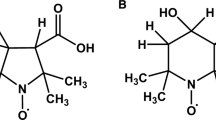
Understanding of electron spin relaxation times is important for predicting feasibility of pulsed electron paramagnetic resonance (EPR) experiments and for wise selection of parameters for continuous wave (CW) EPR spectra. Knowledge of spin relaxation times also supports numerous chemical and analytical applications, including dynamic nuclear polarization (DNP). Since many contributions to relaxation in solution depend strongly on molecular motion, understanding of relaxation also elucidates motional properties. Extensive studies of relaxation in solution as a function of temperature, tumbling correlation time and resonance frequency have been performed for symmetrically substituted triarylmethyl radicals (trityls) [1, 2], benzosemiquinones [3–5], and nitroxides [6–8]. These studies have provided substantial insight into relaxation mechanisms. It is proposed that understanding gained from those studies permits plausible assignments of mechanisms for related systems. To test this proposal, X-band relaxation times at ambient temperature are now reported for BDPA (1:1 α,γ-bisdiphenylene-β-phenylallyl), galvinoxyl (2,6-di-tert-butyl-α-(3,5-di-tert-butyl-4-oxo-2,5-cyclohexadien-1-ylidene)-p-tolyloxy), DPPH (2,2-diphenyl-1-picrylhydrazyl) and thianthrene radicals (Fig. 1).

Molecular structures of the radicals studied
BDPA is an unsymmetrical triarylmethyl radical that contains no heteroatoms. It is used in solution to monitor the kinetics of peroxide scavenging [9, 10] and for DNP [11–14]. DPPH and galvinoxyl are used for applications similar to those of BDPA. In solution, DPPH has been widely used for radical scavenging, especially in food science and natural product investigation [10, 15–17]. Similarly, galvinoxyl has been used to assess antioxidant activity of many food products [18–20]. Like BDPA, DPPH [13, 21] and galvinoxyl [14, 22] have been used for DNP enhancement. The thianthrene cation radical is less common, but derivatives have been utilized in organic synthesis [23] and as EPR-active oxidation probes [24]. Nitroxide radicals may be the most ubiquitous class of compounds studied by EPR. Applications range from spin trapping [25] to spin labeling of biomolecules [26]. The nitroxides discussed here are fully deuterated, except for a single hydrogen on the mHCTPO ring, essentially removing any unresolved proton hyperfine splitting. Like nitroxides, semiquinone and trityl radicals have been extensively studied by EPR.
2 Experimental
2.1 Sources of Materials
DPPH, galvinoxyl, and the 1:1 BDPA:benzene complex were purchased from Sigma Aldrich (St. Louis, MO, USA). Solid thianthrene was prepared by Ashley McDaniel in the laboratory of Matthew Shores (Colorado State University).
2.2 Solution Preparation and Concentration Measured by UV–Vis Absorbance
BDPA, DPPH, and galvinoxyl were dissolved in toluene (Acros Organics). BDPA was prepared at several concentrations: 0.7, 2.8, 5.5, and 12.5 μM. The 12.0 µM concentration of the DPPH solution was confirmed by UV–Vis absorbance at 515 nm based on an extinction coefficient of 12,000 M−1 cm−1 [27]. The 2.8 µM concentration of the galvinoxyl solution was confirmed by UV–Vis absorbance at 428 nm using an extinction coefficient of 175,000 M−1 cm−1 [28]. Due to limited solubility thianthrene was dissolved in trifluoroacetic acid (TFA, Mallinckrodt Baker) instead of toluene, at a concentration of 500 μM.
2.3 Removal of O2
Toluene solutions of BDPA, DPPH, and galvinoxyl were contained in quartz tubes with 4 mm outer diameter (o.d.). Samples were degassed via several cycles of freeze–pump–thaw (FPT). In studies of nitroxides or semiquinones in aqueous solution, purging (bubbling) with N2 gas for 1–2 h was sufficient to remove O2 [5, 29]. FPT is difficult to perform for aqueous solutions because water expansion on thawing frequently breaks tubes. However, for a sample of BDPA in toluene, changes in the resolution of the hyperfine splitting in the CW spectrum (Fig. 2) demonstrated that more than 6 h of continuous N2 purge is required to adequately displace dissolved O2. The higher solubility of O2 in toluene than in water may contribute to the inefficiency in removing O2 by purging toluene solutions with N2. The higher volatility of toluene than of water also makes N2 purging less attractive for toluene than for water solutions.

X-band CW EPR spectra for dilute toluene solutions of BDPA in Teflon tubing with 0.3 mm wall thickness after various time of purging with N2 gas: 2 h, 3 h, 6 h, and 8 h. Y-axis amplitudes are normalized. The decrease in S/N from 6 to 8 h indicates that the radical concentration decreased during this time. Spectra were averaged for 9 scans and collected using 30 kHz modulation frequency, 0.2 G modulation amplitude, and 2 mW microwave power
Solutions of thianthrene in TFA were drawn into thin-walled Teflon tubing [0.97 inner diameter (i.d.)] that was sealed at both ends with Critoseal™. The Teflon tubing was folded over and supported in a 4 mm o.d. tube alongside a purge line (also Teflon) that delivered N2 to exchange O2 through the walls of the tubing. Samples were purged for several hours until T 2 reached a maximum value and remained stable. Purging was continued for the duration of the measurements. FPT preparation was attempted for thianthrene, but sample degradation was rapid under these conditions. Relaxation time measurements were performed within a few hours of thianthrene radical preparation. The linewidths in the spectra of thianthrene radical decrease from about 3.9 G in equilibrium with air to about 0.26 G after oxygen is removed (Fig. 3), which is an unusually large sensitivity to oxygen. By contrast the linewidth for PDT in water at ambient temperature decreases from about 300 mG in equilibrium with air to 160 mG in the absence of oxygen.

X-band CW EPR spectrum of thianthrene radical in trifluoroacetic acid, with concentration <700 μM. Solution purged with N2 (gray). Air saturated solution (black). Spectra were averaged for 4 scans and collected with 100 kHz modulation frequency and 0.02 G modulation amplitude at 0.63 mW microwave power
2.4 EPR Spectroscopy
Continuous wave (CW) EPR to monitor the effect of dissolved oxygen on resolution of small proton hyperfine splittings in the spectra of BDPA (Fig. 2) and on the linewidths for thianthrene (Fig. 3) was performed using a Bruker EMX spectrometer equipped with an ER4122-SHQ (super high quality factor) resonator.
Pulse measurements at X-band (9.5 GHz) were performed on a locally built spectrometer [30] equipped with a Bruker ER4118X-MS5 split ring resonator and a nominal 1 kW TWT amplifier. The resonator was overcoupled to Q ~ 150 to reduce resonator ringdown. T 2 was measured by 2-pulse spin echo decay and T 1 by 3-pulse inversion recovery. Appropriate field positions were determined by recording a field-swept echo-detected spectrum (Figs. 4, 5a) prior to relaxation measurement. Relaxation time measurements were performed at the field position of maximum echo height for BDPA, DPPH, and thianthrene. For galvinoxyl, the high-field line position was used to measure relaxation. The π/2 pulse lengths were 40 ns and power was adjusted to give maximum echo amplitude for each measurement. Relaxation times did not depend on pulse length. The detector gate was set at FWHM for each measurement. Other parameters differed slightly between radicals and were selected to give maximum signal.

X-band field-swept echo-detected spectra demonstrating differences in g values and hyperfine splitting. Amplitudes have been normalized. Spectra were collected using 40 ns π/2 pulse lengths and averaged using multiple scans. The detector gates were selected to maximize S/N, so the resolution was not high enough to reveal all of the hyperfine components

Field-swept echo-detected spectrum (a) and Fourier transform of the FID signal (b) of the thianthrene radical cation. The spectrum in A was collected using a 150 ns π/2 pulse length, and the detector gate was set for optimum resolution
For the thianthrene radical, the free induction decay (FID) is so intense that it is difficult to record a spin echo, even with an iron object placed in the magnetic field to decrease homogeneity. FTEPR measurements were performed at X-band on a Bruker E580 system. The resonator and TWT were comparable to those used on the locally built system, and the resonator was similarly overcoupled to Q ~ 150. The FID was collected using 4-step phase cycling and a 12 ns pulse length. Following acquisition, the data were Fourier transformed and zero-filled to 4,096 points.
2.5 Data Analysis
Relaxation curves were fit with exponential functions using a locally written program, Multifit. It is based on Provencher’s algorithms [31] and compares the quality of fits to single, double, and triple exponentials, assigning a weighting to each component. Both T 2 and T 1 curves fit well to single exponential functions for all samples studied. The resulting relaxation times are listed in Table 1.
2.6 Tumbling Correlation Times
Tumbling-dependent processes make significant contributions to T 1 in fluid solution for many radicals. Tumbling correlation times for trityl-CD3 [1], OX63 [1], PDT [8] and mHCTPO [8] in water and for 2,5-DTBSQ in ethanol [5] have been reported. For small molecules tumbling rapidly in liquid solution, the tumbling correlation time can be estimated by adding an adjustable slip parameter to the Stokes–Einstein calculation such that τ R = c slip V η/kT where V is the molecular volume, η is solvent viscosity, and k is Boltzmann’s constant [32, 33]. The adjustable parameter c slip ranges from 0 to 1.0 depending on the size of the molecule and the extent of solute–solvent interaction [32, 33]. To permit estimates of the contribution of spin rotation to relaxation for BDPA, galvinoxyl, DPPH, and thianthrene in toluene, values of τ R in Table 2 were estimated assuming c slip ~ 0.20. This is higher than for tempone in toluene because of the expectation of significant π–π interaction between the aromatic solutes in toluene, but smaller than the c slip = 0.4 observed for negatively charged semiquinones in alcohol solution [5] or the 0.65 for trityl-CD3 or OX63 in water [1]. Calculations of τ R (Table 2) used the following viscosities (η) at 295 K: toluene 0.55 cP, TFA 0.58 cP, water 1.0 cP, and ethanol 1.08 cP [34].
3 Results and Discussion
3.1 Radical Stability
The following comments concerning radical stability are based on observation for solutions prepared with reagent grade solvents. Additional solvent purification was not attempted. Degradation of the thianthrene cation radical, monitored by UV–Vis absorbance, was rapid in the first 24 h following sample preparation, but then slowed. To obtain reproducible results, fresh samples were prepared each day. Significant loss of signal intensity was observed within 24 h for BDPA. Solutions of galvinoxyl and DPPH were more stable than for BDPA. Degradation was slowed by storage in the dark at −20 °C. In contrast, the nitroxide radicals are remarkably stable compounds due to the steric bulk of substituent groups on the carbons adjacent to the N–O moiety.
3.2 Concentration Dependence of Relaxation Time for BDPA in Toluene
Relaxation times reported in Table 1 are at low concentrations, approaching the limiting values. The relaxation rates for the neutral aromatic radicals BDPA, galvinoxyl, and DPPH in toluene solution are strongly concentration dependent (Fig. 6). The T 1 for galvinoxyl in sec-butyl benzene was found to exhibit concentration dependence even at concentrations as low as 0.1 mM [35]. The slope of the plot of 1/T 1 for BDPA in toluene is 6.2 × 107 s−1/mM. By contrast the slope of an analogous plot for OX63 in water is 3.6 × 104 s−1/mM [36]. The smaller concentration dependence of 1/T 1 for OX63 than for BDPA is attributed to the charged carboxylic acid groups, which result in electrostatic repulsions that decrease collision probability. There may also be a tendency for π-stacking of the neutral aromatic radicals in toluene. 1/T 1 for 2,5-DTBSQ and similar semiquinones is approximately independent of concentration in the mM range [3, 5].

Concentration dependence of 1/T 1 and 1/T 2 for BDPA in deoxygenated toluene
3.3 Comparison of Electron Spin Lattice Relaxation Rates for Organic Radicals in Solution
The relaxation times for BDPA, galvinoxyl, and DPPH in toluene and thianthrene in TFA are listed in Table 1 in order of decreasing T 1. Values were measured at concentrations that are low enough that collisions do not dominate relaxation. Comparison values from the literature are included for trityl-CH3, OX63, 2,5-DTBSQ, and two nitroxides. The molar masses are between 170 and 1340 g/mol and tumbling correlation times are between 500 and 9 ps (Table 2), so the radicals are near the fast tumbling limit in which T 1 ~ T 2. The focus of this paper is on T 1. Values of T 2 may be shorter than T 1 due to incomplete motional averaging of g and/or A anisotropy [37]. Studies of electron spin lattice relaxation as a function of temperature and resonance frequency have identified the dominant relaxation mechanisms for trityl-CD3, OX63, semiquinones, and nitroxides. We seek to compare the relaxation rates for BDPA, galvinoxyl, DPPH, and thianthrene (Table 2) with rates for other radicals with known relaxation mechanisms.
3.3.1 Relaxation Mechanisms in Fluid Solution
The spin rotation mechanism (Eq. 1) was proposed to explain early studies [38, 39].
where i = x, y, z and g e is 2.0023.
Values of \(1/T_{1}^{\text{SR}}\) can be calculated based solely on the g values and the tumbling correlation time, τ R and are listed in Table 2.
The local mode mechanism of relaxation (Eq. 2) was initially proposed for glasses and solid lattices [40], but the absence of a change in slope for plots of 1/T 1 vs. temperature at the melting/softening point indicated that this is the dominant contribution to spin lattice relaxation at X-band for trityl-CH3 and OX63 in liquid solution [2].
where Δloc is the energy of the local mode in Kelvin and C local is determined experimentally.
Studies of the frequency dependence of spin–lattice relaxation for symmetrically substituted triarylmethyl radicals [1], benzosemiquinones in alcohol solutions [5], and nitroxide radicals in water [8, 41, 42] have demonstrated the significance of additional relaxation mechanisms that depend on τ R. These processes modulate anisotropic interactions—g anisotropy (Eq. 3), hyperfine (A i ) anisotropy (Eq. 5), or dipolar coupling to solvent nuclei (Eq. 6).
The contribution to relaxation due to modulation of g anisotropy is given by [6, 43],
where Δg = g zz − 0.5(g xx + g yy ), δg = 0.5(g xx − g yy ), μ B is the electron Bohr magneton and J(ω) is the Bloembergen, Purcell, Pound (BPP) spectral density function (Eq. 4).
where τ R is the tumbling correlation time and ω is the resonance frequency in angular units. At X-band ω ~ 6 × 1010 s−1 so for τ R > 17 ps, ωτ R > 1. Values of 1/T g1 (Table 2) are much smaller than the experimental values of 1/T 1 for the radicals studied.
Modulation of anisotropic coupling to a nucleus with nuclear spin I is given by [6, 7, 43],
Equation (5) is not directly applicable to multiple nuclei. However, it indicates qualitatively that this contribution will (1) increase with the square of the anisotropy of the nuclear hyperfine coupling, (2) increase with increasing I, and (3) will increase as τ R decreases within the relevant motional regime.
Modulation of dipolar coupling to solvent nuclei is given by [1]
where τ solvent is the correlation time for motion of the solvent relative to the radical, and C solvent is a function of the dipolar interaction with solvent nuclei. This contribution has been found to be more significant at resonance frequencies lower than X-band [1, 5].
3.3.2 Trends in Electron Spin Relaxation Rates
Of the radicals for which data are available for comparison in liquid solution, the slowest spin lattice relaxation rates at X-band and ambient temperature are for trityl-CH3 and OX63 [40] (Table 2). The tumbling correlation times (300 and 500 ps, respectively) are relatively long, and g values are close to 2.0023, so the contribution from spin rotation (Eq. 1) is small. It is more than an order of magnitude smaller than the observed relaxation rate. Relaxation rates were measured for the central line in the trityl spectrum in which all carbons are 12C (I = 0). Proton hyperfine couplings are very small, so there is negligible contribution to relaxation from modulation of nuclear hyperfine couplings (Eq. 5). The dominant contribution to relaxation at X-band has been shown to be from a local mode [1, 2].
BDPA is an unsymmetrical triarylmethyl radical, for which 1/T 1 in toluene is about 30 % larger than for trityl-CH3 or OX63 in water. Due to the smaller molar mass of BDPA, the lower viscosity of toluene than of water, and a smaller slip coefficient, the τ R of BDPA in toluene is expected to be about a factor of 10 shorter than for OX63 in water. Even with this large decrease in τ R the spin rotation contribution to relaxation, \(1/T_{1}^{\text{SR}}\), is calculated to be an order of magnitude smaller than the experimental value of 1/T 1 for BDPA. The contribution to relaxation from a local mode depends on the energy of the mode (Δloc) and on a coefficient C loc. The structure of BDPA is substantially different than that of trityl-CH3 or OX63, so it is plausible that changes in either or both of Δloc or C loc are large enough to account for the slightly enhanced relaxation for BDPA. Individual proton hyperfine couplings in BDPA are relatively small, but the approximately equal couplings to eight protons results in extensive splitting of the signal (Fig. 2). Although contributions to relaxation analogous to Eq.5 depend on the square of the hyperfine anisotropy, modulation of the numerous nuclear hyperfine interactions may contribute to enhanced relaxation.
For oxygen-centered benzosemiquinones and galvinoxyl, spin–orbit coupling results in g values that are substantially higher than 2.0023 (Table 2). Due to the relatively short τ R (40 ps) and higher g values, spin rotation makes a significant contribution to spin relaxation for 2,5-DTBSQ (Table 2), that is comparable to the contribution from the local mode [5]. For galvinoxyl in toluene τ R (23 ps) is expected to be smaller than for 2,5-DTBSQ in ethanol which compensates for slightly smaller g values and predicts that the contribution from spin rotation for galvinoxyl in toluene will be similar to that for 2,5-DTBSQ in ethanol. 1/T 1 for galvinoxyl in toluene is about a factor of 3 larger than for 2,5-DTBSQ in ethanol. The substantial increase in relaxation rate suggests a contribution from an additional process. In galvinoxyl there is a substantial proton hyperfine coupling to the unique proton on the central carbon [44]. It is proposed that the substantially enhanced relaxation for galvinoxyl compared with 2,5-DTBSQ is due to modulation of the anisotropic proton hyperfine coupling.
Because the molecular weights are similar, the τ R for DPPH in toluene is expected to be similar to that for galvinoxyl in toluene. However, the g values are not as high for DPPH as for the semiquinones, so spin rotation is a less effective relaxation mechanism for DPPH than for the semiquinones. For DPPH there are large anisotropic hyperfine couplings to two nitrogens [45]. Modulation of these interactions by tumbling is proposed to be the primary reason that 1/T 1 for DPPH is substantially larger than for 2,5-DTBSQ.
The sulfur-centered thianthrene radical has larger spin–orbit coupling than the carbon or oxygen-centered radicals, which results in higher g values. The lower molecular weight makes τ R smaller. Because of the higher g values and smaller τ R, the spin rotation contribution to relaxation for thianthrene is the largest of the radicals included in this study. Within the uncertainty of estimating τ R, spin rotation is estimated to make a substantial contribution to 1/T 1. Modulation of the significant hyperfine couplings to four protons also is likely to be a significant contribution.
Of the radicals included in this study, the nitroxides have the shortest T 1. For rapidly tumbling nitroxides at X-band the spin rotation contribution is comparable to the contribution from modulation of the anisotropic nitrogen hyperfine coupling, and the relative importance is strongly dependent on τ R [29].
4 Conclusions
For trityl radicals that have g values close to 2.0023 and very weak proton hyperfine couplings, 1/T 1 is dominated by a local mode. Increasing spin orbit coupling in radicals containing heteroatoms, including semiquinones, thianthrene, and nitroxide radicals increases g values and increases the contributions to relaxation from spin rotation. Modulation of anisotropic nuclear hyperfine interactions makes a large contribution to relaxation for DPPH and for nitroxyl radicals.
References
R. Owenius, G.R. Eaton, S.S. Eaton, J. Magn. Reson. 172, 168–175 (2005)
A.J. Fielding, P.J. Carl, G.R. Eaton, S.S. Eaton, Appl. Magn. Reson. 28, 239–249 (2005)
S.K. Rengan, M.P. Khakhar, B.S. Prabhananda, B. Venkataraman, Pramana 3, 95–121 (1974)
V. Kathirvelu, H. Sato, S.S. Eaton, G.R. Eaton, J. Magn. Reson. 198, 111–120 (2009)
H.B. Elajaili, J.R. Biller, S.S. Eaton, G.R. Eaton, J. Magn. Reson. (2014, submitted)
B.H. Robinson, C. Mailer, A.W. Reese, J. Magn. Reson. 138, 199–209 (1999)
C. Mailer, R.D. Nielsen, B.H. Robinson, J. Phys. Chem. A 109, 4049–4061 (2005)
J.R. Biller, H. Elajaili, V. Meyer, G.M. Rosen, S.S. Eaton, G.R. Eaton, J. Magn. Reson. 236, 47–56 (2013)
R.C. Lamb, J.G. Pacifici, P.W. Ayers, J. Am. Chem. Soc. 87, 3928–3935 (1965)
V. Butkovic, L. Klasinc, W. Bors, J. Agric. Food Chem. 52, 2816–2820 (2004)
L. Lumata, S.J. Ratnakar, A. Jindal, M. Merritt, A. Comment, C. Malloy, A.D. Sherry, Z. Kovacs, Chem. Eur. J. 17, 10825–10827 (2011)
O. Haze, B. Corzilius, A.A. Smith, R.G. Griffin, T.M. Swager, J. Am. Chem. Soc. 134, 14287–14290 (2012)
J.Z. Hu, J.W. Zhou, B.L. Yang, L.Y. Li, J.Q. Qiu, C.H. Ye, M.S. Solum, R.A. Wind, R.J. Pugmire, D.M. Grant, Solid State Nucl. Mag. 8, 129–137 (1997)
H.E. Kirimli, A. Peksoz, Mol. Phys. 109, 337–350 (2011)
M. Espinoza, C. Olea-Azar, H. Speisky, J. Rodriguez, Spectrochim. Acta A 71, 1638–1643 (2009)
I. Essaidi, Z. Brahmi, A. Snoussi, H.B. Koubaier, H. Casabianca, N. Abe, A. El Omri, M.M. Chaabouni, N. Bouzouita, Food Control 32, 125–133 (2013)
M.F. Ramadan, L.W. Kroh, J.T. Morsel, J. Agric Food Chem. 51, 6961–6969 (2003)
P.T. Gardner, D.B. McPhail, C.G. Duthie, J. Sci. Food Agric. 76, 257–262 (1998)
D.B. McPhail, P.T. Gardner, G.G. Duthie, G.M. Steele, K. Reid, J. Agric. Food Chem. 47, 1937–1941 (1999)
V. Papadimitriou, T.G. Sotiroudis, A. Xenakis, N. Sofikiti, V. Stavyiannoudaki, N.A. Chaniotakis, Anal. Chim. Acta 573, 453–458 (2006)
L. Lumata, M. Merritt, C. Khemtong, S.J. Ratnakar, J. van Tol, L. Yu, L.K. Song, Z. Kovacs, RSC Adv. 2, 12812–12817 (2012)
L.L. Lumata, M.E. Merritt, C.R. Malloy, A.D. Sherry, J. van Tol, L.K. Song, Z. Kovacs, J. Magn. Reson. 227, 14–19 (2013)
H.J. Park, M.S. Park, T.H. Lee, K.H. Park, J. Heterocyclic Chem. 50, 663–667 (2013)
H. Goldberg, I. Kaminker, D. Goldfarb, R. Neumann, Inorg. Chem. 48, 7947–7952 (2009)
D.G. Mitchell, G.M. Rosen, M. Tseitlin, B. Symmes, S.S. Eaton, G.R. Eaton, Biophys. J. 105, 338–342 (2013)
L.J. Berliner, J. Grunwald, H.O. Hankovszky, K. Hideg, Anal. Biochem. 119, 450–455 (1982)
A.H. Ewald, Trans. Faraday Soc. 55, 792–797 (1959)
R.R. Lembke, L.V. Natarajan, R.R. Kuntz, J. Photochem. 21, 157–166 (1983)
J.R. Biller, V. Meyer, H. Elajaili, G.M. Rosen, J.P.Y. Kao, S.S. Eaton, G.R. Eaton, J. Magn. Reson. 212, 370–377 (2011)
R.W. Quine, G.R. Eaton, S.S. Eaton, Rev. Sci. Instrum. 58, 1709–1723 (1987)
S.W. Provencher, J. Chem. Phys. 64, 2773–2777 (1976)
R.E.D. McClung, D. Kivelson, J. Chem. Phys. 49, 3380–3391 (1968)
D. Kivelson, P. Madden, Annu. Rev. Phys. Chem. 31, 523–558 (1980)
A.J. Gordon, R.A. Ford, in The Chemist’s Companion: A Handbook of Practical Data, Techniques, and References (Wiley-Interscience Publication, New York, 1972), pp. 4–5
J. Huisjen, J.S. Hyde, Rev. Sci. Instrum. 45, 669–675 (1974)
B. Epel, M.K. Bowman, C. Mailer, H.J. Halpern, Magn. Reson. Med. 70 (2013). doi:10.1002/mrm.24926
J.H. Freed, in Spin Labeling: Theory and Applications, ed. by L.J. Berliner (Academic Press, New York, 1976), pp. 53–132
P.W. Atkins, D. Kivelson, J. Chem. Phys. 44, 169–174 (1966)
L.T. Muus, P.W. Atkins, Electron Spin Relaxation in Liquids (Plenum Press, 1972)
J.G. Castle Jr, D.W. Feldman, J. Appl. Phys. 36, 124–128 (1965)
J.R. Biller, V.M. Meyer, H. Elajaili, G.M. Rosen, S.S. Eaton, G.R. Eaton, J. Magn. Reson. 225, 52–57 (2012)
J.S. Hyde, J.-J. Yin, W.K. Subczynski, T.G. Camenisch, J.J. Ratke, W. Froncisz, J. Phys. Chem. B 108, 9524–9529 (2004)
B.H. Robinson, D.A. Haas, C. Mailer, Science 263, 490–493 (1994)
B. Kirste, H. Kurreck, M. Sordo, Chem. Ber. 118, 1782–1797 (1985)
S.V. Kolaczkowski, J.T. Cardin, D.E. Budil, Appl. Magn. Reson. 16, 293–298 (1999)
L. Lumata, Z. Kovacs, A.D. Sherry, C. Malloy, S. Hill, J. van Tol, L. Yu, L. Song, E. Merritt, Phys. Chem. Chem. Phys. 15, 9800–9807 (2013)
K. Watanabe, J. Yamauchi, H. Ohya-Nishiguchi, Y. Deguchi, K. Ishizu, Bull. Inst. Chem. Res., Kyoto Univ. 53 (1975)
M. Bennati, C.T. Farrar, J.A. Bryant, S.J. Inati, V. Weis, G.J. Gerfen, P. Riggs-Gelasco, J. Stubbe, R.G. Griffin, J. Magn. Reson. 138, 232–243 (1999)
K. Mukai, T. Tamaki, B. Chem, Soc. Jpn. 50, 1239–1244 (1977)
A.Y. Bresgunov, A.A. Dubinsky, O.G. Poluektov, A.I. Prokof’ev, Mol. Phys. 75, 1123–1131 (1992)
C. Marti, J. Irurre, A. Alvarezlarena, J.F. Piniella, E. Brillas, L. Fajari, C. Aleman, L. Julia, J. Org. Chem. 59, 6200–6207 (1994)
J.S. Hwang, R.P. Mason, L.-P. Hwang, J.H. Freed, J. Phys. Chem. 79, 489–511 (1975)
H. Sato, V. Kathirvelu, A.J. Fielding, S.E. Bottle, J.P. Blinco, A.S. Micallef, S.S. Eaton, G.R. Eaton, Mol. Phys. 105, 2137–2151 (2007)
Yu.V. Yablokov, Dokl. Akad. Nauk SSSR 133, 424–426 (1960)
Acknowledgments
Partial support of this research by NIH grants P41EB002034 (H. J. Halpern, PI) and R01EB00557 and by the University of Denver is gratefully acknowledged. We thank Ashley McDaniel (CSU) for providing the thianthrene and Ralph Weber (Bruker BioSpin) for assistance in obtaining the FTEPR spectrum of the thianthrene radical.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Meyer, V., Eaton, S.S. & Eaton, G.R. X-band Electron Spin Relaxation Times for Four Aromatic Radicals in Fluid Solution and Comparison with Other Organic Radicals. Appl Magn Reson 45, 993–1007 (2014). https://doi.org/10.1007/s00723-014-0579-6
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-014-0579-6