
Abstract
Hepatocellular carcinoma (HCC) is one of the most common cancers associated with high mortality rate. Understanding of events leading to HCC pathophysiology is essential for its better management. We earlier reported development of a novel rodent model by administrating chemical carcinogens, DEN, and 2-AAF for study of HCC at very early stage. 2D-Electrophoresis analysis of total serum proteins identified several differentially expressed proteins in animals undergoing tumorigenesis. MALDI-TOF-MS/MS analyses were performed to characterize the differentially expressed proteins. Further real-time PCR analyses were taken place to quantify the transcript expression for the identified proteins at HCC initiation and tumor stages. Considering protein-protein interactions among the experimentally identified proteins and their interacting neighbors, a protein network has been analyzed that provided further insight into molecular events taking place during HCC development. Histological changes confirmed HCC initiation and hepatotumorigenesis at 1 and 4 months post carcinogen treatment, respectively. Four differentially expressed proteins were identified which were further characterized as regulator of G protein signaling 1 (RGS1), sepiapterin reductase (SPR), similar to zinc finger and BTB domain–containing protein 21 isoform X2 (ZNF295), and a hypothetical protein CXorf58 homolog. Quantification of transcripts for these proteins revealed elevation in their expression both at initiation and tumorigenesis stages. The study deciphers the regulatory role of these proteins during HCC progression.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is globally the fifth most common cancer with very high death rate among cancer patients (Ranjpour et al. 2020). HCC is one of the predominant cancers with different etiologies (Zhen et al. 2017). Several chronic liver diseases may lead to HCC development (van Meer et al. 2013). A number of factors including inflammation (Mossanen et al. 2019), genetic aberrations, and changed expression of regulatory proteins alter biologic pathways during carcinogenesis (P Katare et al. 2018). Patients are usually diagnosed only at advanced stage of HCC when disease management becomes relatively more difficult. Therefore, there is an urgent need for development of new biomarkers for its early detection (Faraj Shaglouf et al. 2020).
The present study has been designed to relatively quantify the mRNA expression of the differentially expressed proteins and get an insight into their regulatory role in HCC initiation and tumorigenesis stages. Furthermore, a HCC-specific network has been generated by analyzing the interaction of global proteins and the proteins deduced from these transcripts. The analysis deciphers regulatory pathways that play important role during HCC development.
Material and methods
Animal model and HCC induction
The study was performed in accordance with the ethical standards. Male Wistar rats weighing 80–100 g were divided into four groups containing six animals each, namely, control (C) and carcinogen-treated groups 1M (sacrificed at 1 month after carcinogen treatment), 4M (sacrificed at 4 months after carcinogen treatment), and 6M (sacrificed at 6 months after carcinogen treatment). Liver cancer was chemically induced in treated groups as reported by us earlier (Malik et al. 2013a). Briefly, DEN (200 mg/kg body weight) and 2-AAF dissolved in 1% carboxymethyl cellulose (150 mg/kg body weight) were used as initiator and promoter of HCC, respectively. A single i.p. dose of DEN was followed by administration of 2-AAF. The first dose of 2-AAF was given 1 week after DEN administration. Three doses of 2-AAF were orally administered on three alternative days of the first week of each month, for 4 months; thus, the treated-group 1M received 3 doses of 2-AAF while the treated-group 4M and 6M received 12 doses of 2-AAF. The controls were sham treated with normal saline at the same schedule. Rats were anesthetized with diethyl ether, sacrificed at the end of 1 month (1M group, initiation stage), 4 months (4M group, tumorigenesis stage), and 6 months (6M group, tumorigenesis stage), respectively, and livers were excised for further analysis.
Histological examination
Livers were fixed in formalin (10%) for 24 h, sliced (3–4 mm), dehydrated, and embedded in paraffin. Four cross-sections were taken from each liver and stained with hematoxylin and eosin followed by two washings with xylene. Sections were mounted with DPX mountant and observed under light microscope for histological changes. The histological examination confirmed HCC induction at 1 month after carcinogen treatment and tumorigenesis at 4 months after carcinogen treatment when fully grown tumors were observed. The details of histopathology analysis have been reported by us earlier (Ranjpour et al. 2018a).
Preparation of serum sample for proteome profiling
Rats were anesthetized and blood was drawn from their tail vein. Blood samples were allowed to stand at room temperature for 30 min and the coagulated blood was centrifuged at 1685×g for 20 min; serum was separated and stored at − 70 °C.
High abundance protein depletion
Serum samples were selectively depleted of albumin and IgG using GeNei albumin and IgG depletion kit using manufacturer’s protocol. Gradient SDS-PAGE with 12% acrylamide gel was performed to compare the serum protein profiles of control vis-a-vis HCC-bearing animals.
2D-Electrophoretic analysis of proteins
The protein concentration was quantified by Bradford’s method (Bradford 1976). Aliquots containing 100 μg proteins were diluted to 200 μl in rehydration buffer. The 11 cm, pH 4-7 IPG strips (GE Healthcare, ImmobilineTM DryStrip) were incubated with rehydrated protein samples for 16 h and focused using IEF apparatus (EttanIPGphor 3 GE Heathcare) for 500 V-1 h, 1000 V-1 h, 6000 V-2 h, and 6000 V for a total of 35,000 V-hour. The strips were washed and equilibrated with buffer 1 (2% DTT, 6M urea, 75 mM Tris-HCl, pH 8.8, 2% SDS, 29.3% glycerol, and 0.002% bromophenol blue) followed by equilibration with buffer 2 (2.5% IAA, 6M urea, 75 mM Tris-HCl, pH 8.8, 2% SDS, 29.3% glycerol, and 0.002% bromophenol blue) for 15 min each. SDS-PAGE was performed using SE 600 Ruby electrophoresis unit (GE Healthcare). The equilibrated strips were placed on 12% poly-acrylamide gel and electrophoresis was carried out at 120 V using 190 mM Tris-glycine buffer, pH 8.3. The separated proteins were silver stained and differentially expressed protein spots were identified using PD-Quest software. The protein spots of interest were excised from the gel for further analysis by MALDI-TOF-MS/MS characterization.
Characterization of the experimentally identified proteins with MALDI-TOF-MS/MS analysis
The excised protein spots were stored in 5% glacial acetic acid, washed with Milli-Q water, and incubated with Farmer’s reagent (200 μl). The gel pieces were then put in 200 μl of 50:50 (v/v) mixture of 25 mM ammonium bicarbonate: acetonitrile (ACN) and shaken on a rocking platform for 20 min. After discarding the supernatant, gel pieces were treated with 100% ACN for complete dehydration. After ACN evaporation, gel pieces were rehydrated with 20 mg/ml trypsin prepared in 25 mM ammonium bicarbonate and incubated overnight at 37 °C. The reaction was stopped by adding 1% formic acid. The supernatant containing tryptic peptides was collected, mixed with 50% ACN and 0.1% trifluoroacetic acid (TFA), and incubated for 5 min at room temperature. The extraction was repeated with 0.5% TFA and samples were stored at − 20 °C. For further analysis, the samples were reconstituted in a solution containing ACN (50%) and TFA (0.1%). CHCA matrix (2.5 μl) was added to 2.5 μl of samples and 1 μl was spotted on a MALDI-TOF plate. Analysis was carried out using MALDI-TOF/TOF Ultraflex III instrument. The following search parameters were selected: peptide mass tolerance of ± 200 ppm, fragment mass tolerance of ±0.4 Da, peptide charge of 1+. Peptides were then identified by database search against the MASCOT search. The hit significant (p < 0.05) in accordance with Mascot Probability Search was only accepted and the max missed cleave was taken as 1 (Ranjpour et al. 2018a).
Relative quantification of transcript expression by real-time PCR analysis
Total RNA was isolated from the liver using RNA extraction kit (RNA Sure Mini kit, cat.#NP.84105) and concentration was measured by Nanodrop Spectrophotometer (NanoDrop 2000C, Thermo Scientific). The quality of RNA was checked on 1.2% denaturing agarose gel. The absence of genomic DNA contamination was confirmed by PCR with β-actin primers targeted for 1234 bp amplicon (representing an intron in β-actin gene). Absence of amplification band confirmed that RNA preparation was free of DNA contamination. One microgram of normalized RNA was reverse transcribed using RevertAidTM H Minus, First Strand cDNA Synthesis kit (Fermentas) and the integrity of cDNA was checked by PCR using β-actin primers, specific for a 207 bp target on the cDNA. For PCR amplification, the reaction mixture (25 μl) contained 1 μl of cDNA, 12.5 μl of 2X Dream Taq PCR Master mix (Thermo Scientific), and 0.4 μM of each primer. The conditions for PCR amplification were initial denaturation at 95 °C for 5 min; 35 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 45 s, extension at 72 °C for 45 s, followed by final extension at 72 °C for 5 min. The reaction products were analyzed by electrophoresis on 1.5% agarose gel in 1X TAE buffer system. Quantitative PCR was performed in total volume of 25 μl using 12.5 μl of 2X SYBR green master mix (Maxima), 0.4 μM of each primer, 1 μl of 5 times diluted cDNA, and 9.5 μl of nuclease free water. All reactions were performed in triplicate on the Stratagene Mx3005P Q-PCR System (Agilent Technologies, USA). The β-actin gene was used as reference normalizer and control samples were taken as calibrator. The constant threshold value was used to obtain the Ct values. The relative gene expression was quantified with comparative ΔCt(2−ΔΔCT) method of Livak and Schmittgen (2001). The nucleotide sequences of the primers that were used to amplify the desired transcripts and amplicon sizes are shown in Table 1.
Generation and analysis of protein interaction network
Our group recently identified several differentially expressed proteins using sera of the carcinogen-treated rats which were further characterized by MALDI-TOF-MS/MS analysis (Table 2) (Malik et al. 2013b; Katare et al. 2018; Ranjpour et al. 2018a, b). Using Uniprot database, the IDs for the characterized proteins were found. These proteins along with the differentially expressed proteins introduced by the present study were fed into the STRING software following selection of “multiple proteins” option from the software while the organism option was selected as “Rattus norvegicus.” The maximum numbers of interactors for the first and second shells were selected as “no more than 10 interactors” and the settings of the software were further updated. The network among these proteins and their interacting partners was analyzed. The routes of master network associated with the MALDI-TOF-MS/MS characterized proteins were identified representing the HCC-specific network, in the represented animal model, which depicts interactions among experimentally characterized proteins and their interacting partners for better understanding of HCC at molecular levels. Moreover, GENEMANIA software was used to analyze the potential co-expression and co-localization of the expressed genes coding for the experimentally characterized proteins (Franz et al. 2018).
Statistical analysis
The experiments were conducted in triplicate and the data have been presented as mean ± S.E.M. The significance of differences between control and treated groups was analyzed using one-way ANOVA followed by the Dunnett tests and considered statistically significant when p < 0.05.
Results
HCC induction and histological evaluation
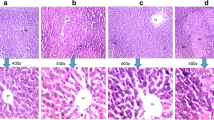
HCC was successfully induced by administrating chemical carcinogens, DEN, and 2-AAF to male Wistar rats (Ranjpour et al. 2018a, b), using the protocol as reported by us earlier (Malik et al. 2013a). The novel critical differences in the proposed model are the elimination of both necrogenic doses of carcinogens and partial hepatoctemy. HCC development was confirmed by histological analyses (Fig. 1). Presence of neoplastic cells revealed as an indicator of liver cancer initiation. The histology showed that there are nodules that are not localized in any specific area but are present all over the liver tissue. This further supports our notion that the tumors have field cancerization origin. Similar field cancerization may be taking place in case of human liver cancers also (Ranjpour et al. 2018a, b). Morphological analysis of the liver also showed development of malignant tumors at 4 months after carcinogen treatment. Analysis showed HCC induction at 1 month after carcinogen treatment and alteration in liver structure and tumorigenesis at four months post carcinogen treatment (Ranjpour et al. 2018a).

Histological analysis of liver tissue. Step-wise histological analysis revealed presence of hemorrhage, multi focal lesions, enlarged vacuolated hepatocytes with clear cytoplasm, and cystic degeneration of hepatocytes with deposition of eosinophilic material from early hyperplasia up to malignant transformation of hepatocytes. a Control showing normal architecture of the liver; HA: hepatic artery; BD: bile duct. b One month after carcinogentreatment. c Four months post carcinogen treatment. d Six months after carcinogen treatment. Nuclear atypia is observed in adenoma in circles. Scale bar is 20 μm at × 400 magnifications
Proteomic studies
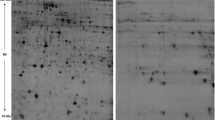
2D-Elecrophoresis analysis of total serum proteins was performed. Step-wise analysis revealed consistent changes in protein expression level associated with HCC progression, starting from 1 month (initiation) up to 6 months (hepatotumorigenesis) post carcinogen treatment; Fig. 2 represents the 2D-profiles of control and carcinogen-treated animals where target proteins have been marked.

Analysis of differentially expressed proteins. Serum proteomic analysis of carcinogen-treated animals and control has been represented using 2D-electrophoresis. The up-regulation in expression of protein spots, namely, R, S, and X, has been observed while there is a newly expressed spot (Z) whose expression has been increased during HCC progression in carcinogen-treated animals (a control, b 1 month, c 4 months, and d 6 months post carcinogen treatment). The elevation in expression of these spots was statistically significant (p < 0.05)
The PD-Quest analysis of 2D-gels was carried out that analyzed the differentially expressed proteins at HCC initiation and tumorigenesis stages. Differences in protein spot area and intensity revealed changed expression of these proteins. The analysis assigned unique sample spot protein (SSP) numbers to protein spots and revealed up-regulation in expression of several proteins out of which four, namely, R, S, X, and Z, were randomly selected in which elevated expression was consistently observed throughout HCC progression. The analysis quantified the intensity of the target proteins representing their expression in HCC initiation and tumorigenesis stages (Fig. 3).

PD-Quest analyses of 2D-gels. The expression of the target proteins on 2D-gels has been quantified using PD-Quest analysis. The master gel has protein spots from both control (C) and treated groups. The pop-up graphs of target spots (R, S, X, and Z) are noted in the boxes comparing protein expression based on intensity (INT) × area between control and treated groups. The unique SSP numbers for protein spots have been written below the analyzed spots. a Protein spots and their quantified intensities at one month (1M) after carcinogen treatment. The protein marker loaded on the right side of the gel shows the approximate experimental molecular weight of spots. b Protein spots and their quantified intensities at four months (4M) after carcinogen treatment. c Protein spots and their quantified intensities at six months (6M) post carcinogen treatment
The target spots denoted as R, S, X, and Z were characterized by MALDI-TOF-MS/MS analysis as regulator of G protein signaling 1 (RGS 1), sepiapterin reductase (SPR), putative CXorf58 homolog, and similar to zinc finger and BTB domain–containing protein 21 isoform X2 (ZNF295), respectively. The SSP numbers of the characterized proteins assigned by PD-Quest analysis and their intensities at 1 (1M), 4 (4M), and 6 (6M) months after carcinogen treatment along with their peptide sequences characterized by MALDI-TOF-MS/MS analysis and their approximate experimental molecular weight have been shown in Tables 3 and 4.
Real-time PCR analysis to quantify the transcript expression
The real-time PCR analysis relatively quantified the transcript expression for the denoted proteins, at HCC initiation and tumorigenesis stages. The analysis for RGS1 mRNA expression revealed 16-fold increase at initiation stage, that further increased to 63-fold at 4 months (tumorigenic state) but went down to 17-fold at 6 months post carcinogen treatment vis-a-vis control. The expression of mRNA for protein CXorf58 homolog was increased by 57-fold at 1 month, continued to increase further to 80- and 136-fold at 4 and 6 months after carcinogen treatment, respectively. Similarly, the real-time PCR analysis for expression of SPR mRNA revealed 18-fold increase at 1 month when it got stabilized and was 17- and 18-fold up-regulated at 4 and 6 months after carcinogen treatment, respectively. The expression of mRNA for zinc finger and BTB domain–containing protein 21 isoform X2 showed a continued increase that revealed 88-fold increase at 1 month after carcinogen treatment and 192- and 225-fold elevation at 4 and 6 months after carcinogen treatment. The representative data of real-time analysis have been shown in Fig. 4.

Real-time analysis of transcripts for the target proteins. The transcript expression for the characterized proteins has been relatively quantified using real-time PCR analysis. The analysis showed elevation in mRNA expression of a RGS 1, b putative protein cxorf 58 homolog, c SPR, and d zinc finger and BTB domain–containing protein 21 isoform X2 (ZNF 295). Fold change in expression of carcinogen-treated transcripts were calculated over age-matched controls. Data presented as mean ± SEM (n = 3). p < 0.0001 using one-way ANOVA followed by the Dunnett test
Protein interaction study
We have analyzed a network of HCC-associated proteins involving experimentally identified proteins and their interactors.
The network revealed interactions among SPR and RGS1 through AKT/MTOR and GSK3B. SPR is connected to MTOR through AKT; however, the interrelationship among RGS1 and MTOR is mediated through GSK3B and AKT, although RGS1 can also be connected to MTOR through GSK3B, directly. The direct interactions among ARAF and ApoA1, TTR and ApoA1, and C3 and ApoA1 have been shown by the analysis revealing ApoA1 as the mediator among ARAF, TTR, and C3. Further ARAF connects the ApoA1/TTR/C3 complex to AKT/MTOR. On the other hand, AKT is directly connected to ARAF and ApoA1 and can therefore connect ARAF and ApoA1/TTR/C3 complex to SPR and RGS1 through GSK3B. The interaction among SPR and ARAF has also been mediated through AKT (Fig. 5).

HCC-associated protein network. The STRING software was used to analyze the proposed network. The segregated network introduces AKT and ARAF as the hub among the experimentally characterized proteins connecting several proteins together. The experimentally characterized proteins are shown within the red circles
Gene network analysis using GENEMANIA
The analysis revealed that spr gene is co-expressed along with genes coding for araf, c3, camk2g, and map2k2 whereas, the rgs1 gene is co-expressed with c3 and tsc22d1 genes. The network also showed that the spr gene is co-localized with araf and c3 genes while rgs1 gene gets co-localized with c3 and tsc22d1 genes (Fig. 6).

Potential gene expression during HCC progression. The network has been analyzed using GENEMANIA software. The potential co-expression and co-localization of the target genes have been shown in the figure. The roles associated with candidate genes coding for the experimentally identified proteins have been shown during HCC development. CAMK2 phosphorylates SPR and plays important role in its activation and thus regulates tyrosine formation catalyzed by SPR (Ranjpour et al. 2018a). TSC22D1 is a target for TGF-β gene (Rentsch et al. 2006) and interacts with p53, protecting it from poly-ubiquitination and enhances its expression (Yoon et al. 2012). The genes coding for the target proteins characterized in the present study (SPR and RGS1) are shown within the blue circles. The genes coding for proteins characterized by our group, previously, are shown in red circles
Discussion
The study highlights the results on animal model and co-relates these observations with available information in literature. We reviewed the molecular biology of premalignancy and early lesions in a specific model developed for HCC that could support us to introduce several proteins in which dysregulated transcription and translation have regulatory roles for early detection of HCC. Using bioinformatic tools, we analyzed a HCC-specific network among these differentially expressed proteins and their interacting partners associated with signaling pathways to better understand the molecular mechanisms taking place during HCC progression in order to introduce potential diagnostic or/and prognostic targets for HCC management and overcome the death rate associated with its detection at late stages.
Liver cancer was chemically developed in treated groups as reported by us earlier (Malik et al. 2013a). The model was successfully reproduced in the present study (Ranjpour et al. 2018a, b, 2020). The blood samples were collected from the treated and control groups. The serum was stored at − 80° deep freezer. Then, the animals were sacrificed and different sections from the liver were sent for histopathology analyses keeping the adjacent sections in RNAlater or sometimes kept frozen for RNA isolation. The chemically induced carcinogenesis in our model was confirmed by histopathology evaluation. The details have been published earlier. Based on analysis of the histology data, we have reasons to believe that the tumors have been developed as the result of field cancerization and are not of monoclonal origin. Similar field cancerization may be taking place in case of human liver cancers also. Due to technical reasons, micro-dissection was not used to have cancer tissue and non-cancer tissues from the same animal. We used tissues from carcinogen-treated and placebo-treated animals as the experimental and controls in our study. However, as the study reports the changes in expression of genes of interest in HCC over the control, this strategy should not change the qualitative observations of our study. The progression of liver cancer was monitored by analysis of morphological and histological parameters during HCC development. Histological evaluation showed characteristic features of neoplastic cells and confirmed the time-bound progression of HCC (Ranjpour et al. 2018a). After the histopathology reports were received, the serum and adjacent tissue sections were used for 1D, 2D, and RT-PCR, accordingly. The serum protein profile of control and carcinogen-treated rats were compared using 1D- and 2D-electrophoresis. The changes in expression of HCC-related proteins were analyzed starting from initiation of liver cancer and at hepatotumorigenesis. The differentially expressed proteins were identified by PD-Quest analysis of 2D-gels; some of these proteins were later characterized using MALDI-TOF-MS/MS analysis. The expression level of the target proteins and the mRNA coding for these were quantified at HCC initiation and during tumorigenesis stages that revealed translation and transcription analyses complement each other. The differentially expressed proteins characterized in this study play important role in pathophysiology of HCC.
RGS1 was found to be up-regulated in the present study and as per our review its up-regulation has not so far been reported during early stages of HCC. RGS1 stimulates proliferation, invasion, and metastasis in malignancies. The regulatory role of RGS1 has been reported in CXCL12-CXCR4 chemokine signaling in normal plasmablasts and multiple myeloma cell line representing its vital role in pathophysiology of plasma cells and multiple myeloma (Roh et al. 2016). Elevated level of RGS1 increases the GTPase activity of small G proteins and deregulates signal transduction that leads to cell proliferation dysregulation, cancer cell migration, and tumorigenesis (Sethakorn and Dulin 2013). RGS1 has been found to be up-regulated in majority of solid tumors (Sethakorn and Dulin 2013) including melanoma, head and neck squamous cell carcinoma, adult T-cell leukemia, renal cell carcinoma, ovarian cancer, cervical cancer, and mantle cell lymphoma (Hurst and Hooks 2009). The regulator of G protein signaling (RGS) family is a diverse group of multifunctional proteins that regulate cellular signaling events downstream of G protein–coupled receptors (GPCRs) (Hurst and Hooks 2009). In recent years, GPCRs have been linked to the initiation and progression of multiple cancers; thus, regulators of GPCR signaling are also likely to be important to the pathophysiology of cancer (Hurst and Hooks 2009). There has been one report showing low levels of RGS1 in tumors of HBV- and HCV-infected HCC patients at multiple tumor stage (Tsai et al. 2006). It may be due to extensive damage to the liver during metastasis as well as in case of tumor recurrence where loss of liver function has taken place during chemo- and/or radio-therapy and surgical intervention. This can explain the apparent discrepancy between the report and our study showing elevated levels of RGS1, at both mRNA and protein levels, at early stage of HCC initiation and its progression.
Highly elevated expression of putative protein CXorf58 homolog bears direct correlation with HCC initiation and tumorigenesis. However, relatively little information is available in the literature to decipher the precise role of this protein in HCC initiation and/or progression. The elevation in zinc finger and BTB domain–containing protein 21 (ZNF295) expression is supported by a study reporting significant elevation in expression of zinc finger and BTB domain–containing protein 20 (ZBTB20) at both protein and mRNA levels at HCC tissues of human patients as compared to the adjacent non-tumor tissues and normal controls (Wang et al. 2011). A relationship between HCC recurrence rate and higher expression of ZBTB20 has also been reported. The increased ZBTB20 expression has correlation with poor prognosis in HCC patients (Wang et al. 2011). ZNFBTB domains play pivotal role in regulation of subcellular localization and binding of transcription factors, thus, controlling the gene expression. Several ZNFBTB proteins act as transcriptional repressor and suppress down-stream gene expression by recruiting co-repressors and several chromatin modifiers. On the other hand, certain other ZNFBTB domains activate gene transcription by recruiting co-activators. Their over expression has been associated with tumorigenesis (Jen and Wang 2016), metastasis, and recurrence rate in cancers (Baas et al. 2003).
We reported up-regulation in expression of SPR in HCC initiation and at tumor stages. Our finding has been supported by a study that revealed elevated expression of SPR in human HCC that is correlated with poor prognosis, higher metastasis, and shorter survival rates indicating its potential role as a tumor promoter (Wu et al. 2020). On the other hand, animal studies revealed that depletion of SPR inhibits proliferation and promotes apoptosis of cancer cells (Wu et al. 2020). SPR catalyzes the final step in tetrahydrobiopterin (BH4) in tissues. BH4 is a co-factor for phenylalanine hydroxylase that catalyzes formation of tyrosine from phenylalanine in the liver (Haruki et al. 2016). Availability of tyrosine in cellular amino acid pool would in turn regulate protein synthesis. Higher BH4 availability would result in higher synthesis of tyrosine which may modulate the synthesis of required proteins and may facilitate the uncontrolled cell division during HCC development (Ranjpour et al. 2018a), Though it is reported that the enzymatic activity of SPR is not associated with development of HCC (Wu et al. 2020). SPR also regulates the levels of intracellular reactive oxygen species (ROS) and shows oxidoreductase activity that is an important cellular mechanism to overcome oxidative stress (Yoo et al. 2009). ROS-based DNA damage, genomic vulnerability of hepatocytes, and T-lymphocyte suppression assist in HCC development (Ogunwobi et al. 2019). Constant activation of inflammatory pathways leads to elevation in secretion of inflammatory cytokines that increases ROS generation (Ogunwobi et al. 2019). Our group recently reported the increased expression of inflammatory cytokines IL-6 and TNF alpha, in rats at carcinogen-treated groups during HCC progression (Ranjpour et al. 2018b).
The significance of the proposed network is that it has been analyzed based on HCC-associated proteins experimentally identified at very early stage, soon after HCC was chemically induced (at 1 month). Our group recently characterized ARAF, ApoA1 (Ranjpour et al. 2018b), TTR, and complement 3 (C3) (Malik et al. 2013b; Katare et al. 2018) using sera of carcinogen-treated rats and reported elevation in their expression in HCC initiation, progression, and at tumorigenesis stages. The increased expression of these proteins was further validated with human sera of liver cancer patients (Malik et al. 2013b; Ranjpour et al. 2018a, b). These proteins along with the proteins characterized in the present study were taken together to construct the proposed master network. The association of SPR with MTOR/AKT1 signaling pathway has been reported by the represented network that was previously validated experimentally (Feinmesser et al. 1999; Fujimoto et al. 2002; Fuks et al. 2001; Gao et al. 2007; Kusaba et al. 2005; Li et al. 2006; Sarbassov et al. 2005; Shao et al. 2003). The network revealed that ARAF and AKT can be introduced as the mediators correlating ApoA1, TTR, and C3 with SPR and RGS1. AKT1 is co-localized in nucleus and coordinate to regulate the activity of specific transcription factor (Gao et al. 2007). AKT is activated by MTOR and its deregulation is an important factor associated with cancer pathogenesis (Kusaba et al. 2005; Sarbassov et al. 2005). MTOR activates STAT3 (Kusaba et al. 2005) that causes cell growth and inhibits apoptosis (Shao et al. 2003). The network deciphers the correlation among these proteins that regulates synthesis of protein and lipoprotein (SPR), lipogenesis (TTR), and proliferation (RGS1 a regulatory element of MAPK signaling).
Conclusion
The proteins reported in this study play important regulatory roles in the liver and elevation in their expression both at mRNA and protein levels may be one of the key factors in liver dysfunctions and HCC progression. Besides characterizing the HCC-associated proteins and quantifying the elevated expression of their transcripts at early initiation and tumor stages of cancer, the study also reveals correlation among the identified proteins with their interacting neighbors and principal molecules associated with HCC regulation at early initiation and tumor stages. The experimental and bioinformatic data taken together reveal that dysregulation in expression of the experimentally characterized proteins may lead to continuous cell proliferation, cancer cell migration, and tumorigenesis in HCC. Thus, a concept of molecule groups could be introduced for prediction of HCC initiation and tumorigenesis that ultimately lead to metastasis.
References
Baas A, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC (2003) Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J 22:3062–3072
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1016/0003-2697(76)90527-3
Faraj Shaglouf LH, Ranjpour M, Wajid S, Jain SK (2020) Elevated expression of cellular SYNE1, MMP10, and GTPase1 and their regulatory role in hepatocellular carcinoma progression. Protoplasma 257:157–167. https://doi.org/10.1007/s00709-019-01423-w
Feinmesser RL, Wicks SJ, Taverner CJ, Chantry A (1999) Ca2+/calmodulin-dependent kinase II phosphorylates the epidermal growth factor receptor on multiple sites in the cytoplasmic tail and serine 744 within the kinase domain to regulate signal generation. J Biol Chem 274:16168–16173
Franz M, Rodriguez H, Lopes C, Zuberi K, Montojo J, Bader GD, Morris Q (2018) GeneMANIA update 2018. Nucleic Acids Res 46:W60–w64. https://doi.org/10.1093/nar/gky311
Fujimoto K, Takahashi SY, Katoh S (2002) Mutational analysis of sites in sepiapterin reductase phosphorylated by Ca2+/calmodulin-dependent protein kinase II. Biochim Biophys Acta 1594:191–198
Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J 20:2536–2544. https://doi.org/10.1093/emboj/20.10.2536
Gao H, Yu Z, Bi D, Jiang L, Cui Y, Sun J, Ma R (2007) Akt/PKB interacts with the histone H3 methyltransferase SETDB1 and coordinates to silence gene expression. Mol Cell Biochem 305:35–44. https://doi.org/10.1007/s11010-007-9525-3
Haruki H, Hovius R, Pedersen MG, Johnsson K (2016) Tetrahydrobiopterin biosynthesis as a potential target of the kynurenine pathway metabolite xanthurenic acid. J Biol Chem 291:652–657. https://doi.org/10.1074/jbc.C115.680488
Hurst JH, Hooks SB (2009) Regulator of G protein signaling (RGS) proteins in cancer biology. Biochem Pharmacol 78:1289–1297. https://doi.org/10.1016/j.bcp.2009.06.028
Jen J, Wang Y-C (2016) Zinc finger proteins in cancer progression. J Biomed Sci 23:53. https://doi.org/10.1186/s12929-016-0269-9
Katare DP, Malik S, Mani RJ, Ranjpour M, Jain SK (2018) Novel mutations in transthyretin gene associated with hepatocellular carcinoma. Mol Carcinog 57:70–77. https://doi.org/10.1002/mc.22732
Kusaba H, Ghosh P, Derin R, Buchholz M, Sasaki C, Madara K, Longo DL (2005) Interleukin-12-induced interferon-gamma production by human peripheral blood T cells is regulated by mammalian target of rapamycin (mTOR). J Biol Chem 280:1037–1043. https://doi.org/10.1074/jbc.M405204200
Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP (2006) The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J Biol Chem 281:19489–19500. https://doi.org/10.1074/jbc.M513249200
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)). Method Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
Malik S, Bhatnagar S, Chaudhary N, Katare DP, Jain S (2013a) DEN+ 2-AAF-induced multistep hepatotumorigenesis in Wistar rats: supportive evidence and insights. Protoplasma 250:175–183
Malik S, Bhatnagar S, Chaudhary N, Katare DP, Jain SK (2013b) Elevated expression of complement C3 protein in chemically induced hepatotumorogenesis in Wistar rats: a correlative proteomics and histopathological study. Exp Toxicol Pathol 65(6):767–773. https://doi.org/10.1016/j.etp.2012.11.003
Mossanen JC, Kohlhepp M, Wehr A, Krenkel O, Liepelt A, Roeth AA, Möckel D, Heymann F, Lammers T, Gassler N, Hermann J, Jankowski J, Neumann UP, Luedde T, Trautwein C, Tacke F (2019) CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4+ T-cell-dependent control of senescence. Gastroenterology 156(6):1877–1889.e4. https://doi.org/10.1053/j.gastro.2019.01.2472019.01.247
Ogunwobi OO et al (2019) Mechanisms of hepatocellular carcinoma progression. World J Gastroenterol 25:2279–2293. https://doi.org/10.3748/wjg.v25.i19.2279
Ranjpour M, Katare DP, Wajid S, Jain SK (2018a) HCC specific protein network involving interactions of EGFR with A-Raf and transthyretin: experimental analysis and computational biology correlates. Anti Cancer Agents Med Chem. https://doi.org/10.2174/1871520618666180507141632
Ranjpour M, Wajid S, Jain SK (2018b) Elevated expression of A-Raf and FA2H in hepatocellular carcinoma is associated with lipid metabolism dysregulation and cancer progression. Anti Cancer Agents Med Chem. https://doi.org/10.2174/1871520618666181015142810
Ranjpour M, Wajid S, Jain SK (2020) Elevated expression of cytosolic phospholipase A2 delta is associated with lipid metabolism dysregulation during hepatocellular carcinoma. Progression Cell J 22:17–22. https://doi.org/10.22074/cellj.2020.6527
Rentsch CA et al (2006) Differential expression of TGFβ-stimulated clone 22 in normal prostate and prostate cancer. Int J Cancer 118:899–906
Roh J et al (2016) RGS1 expression is associated with poor prognosis in multiple myeloma. J Clin Pathol 70:jclinpath-2016. https://doi.org/10.1136/jclinpath-2016-203713
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–1101. https://doi.org/10.1126/science.1106148
Sethakorn N, Dulin NO (2013) RGS expression in cancer: oncomining the cancer microarray data. J Recept Signal Transduct Res 33:166–171. https://doi.org/10.3109/10799893.2013.773450
Shao H, Cheng HY, Cook RG, Tweardy DJ (2003) Identification and characterization of signal transducer and activator of transcription 3 recruitment sites within the epidermal growth factor receptor. Cancer Res 63:3923–3930
Tsai CC et al (2006) Gene expression analysis of human hepatocellular carcinoma by using full-length cDNA library. J Biomed Sci 13:241–249. https://doi.org/10.1007/s11373-005-9062-6
van Meer S, de Man RA, Siersema PD, van Erpecum KJ (2013) Surveillance for hepatocellular carcinoma in chronic liver disease: evidence and controversies. World J Gastroenterol 19:6744–6756. https://doi.org/10.3748/wjg.v19.i40.6744
Wang Q et al (2011) Zinc finger protein ZBTB20 expression is increased in hepatocellular carcinoma and associated with poor prognosis. BMC Cancer 11:271
Wu Y et al (2020) Sepiapterin reductase promotes hepatocellular carcinoma progression via FoxO3a/Bim signaling in a nonenzymatic manner. Cell Death Dis 11:248–248. https://doi.org/10.1038/s41419-020-2471-7
Yoo DR, Chong SA, Nam MJ (2009) Proteome profiling of arsenic trioxide-treated human hepatic cancer cells. Cancer Genomics Proteomics 6:269–274
Yoon CH et al (2012) Crucial role of TSC-22 in preventing the proteasomal degradation of p53 in cervical cancer. PLoS One 7:e42006. https://doi.org/10.1371/journal.pone.0042006
Zhen C, Zhu C, Chen H, Xiong Y, Tan J, Chen D, Li J (2017) Systematic analysis of molecular mechanisms for HCC metastasis via text mining approach. Oncotarget 8:13909–13916. https://doi.org/10.18632/oncotarget.14692
Funding
The work was supported by a MRP grant from the University Grant Commission (UGC) to S. K. Jain [Grant # 40-155/2011 (SR)]. M. R. is grateful to ICCR, Govt. of India, for their financial support [Grant # 2-261/2013-14/ISD-II].
Author information
Authors and Affiliations
Contributions
All authors have contributed equally and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Experiments were conducted following the guidelines of the “Committee for the Purpose of Control and Supervision of Experiments on Animals” (approval number granted by the ethics committee of Jamia Hamdard University is 908/CPCSEA), Government of India. The study was performed in accordance with the ethical standards.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Jörn Bullerdiek
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ranjpour, M., Wajid, S. & Jain, S.K. Elevated expression of sepiapterin reductase, regulator of G protein signaling 1, hypothetical protein CXorf58 homolog, and zinc finger and BTB domain–containing protein 21 isoform X2 is associated with progression of hepatocellular carcinoma. Protoplasma 258, 1133–1143 (2021). https://doi.org/10.1007/s00709-021-01632-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00709-021-01632-2