
Abstract
A series of some new diorganotin(IV) complexes [R2SnLCl] was synthesized by the reaction of 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione and 4-substituted anilines (p-OCH3, p-NO2, p-CH3, p-Cl) with R2SnCl2, (R=Me, Et, n-Bu, Ph) in 1:1 molar ratio. The structure of the Schiff bases and their complexes were characterized by IR, 13C, 1H, 119Sn NMR, and mass spectral techniques. The synthesized ligands and derived organotin complexes were evaluated in vitro against some bacterial strains, viz., Escherichia coli, Pseudomonas aeruginosa, Bacillus cereus, Staphylococcus aureus and fungal strains, viz., Aspergillus flavus, Aspergillus niger, and Candida albicans by serial dilution method. The antimicrobial results revealed that organotin complexes showed a distinct escalation in biocidal activity. Phenyl and butyl complexes were found to be more intoxicating. Furthermore, we performed QSAR studies which explained the different factors affecting the enhancement in the bioactivity of the complexes.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the last few years, the major problem has arisen due to the expression of resistance by microorganisms against the marketed drugs; therefore, there is a great need for the discovery of innovative efficacious therapeutic drugs with better and new mechanisms against microbes [1,2,3]. To develop potential drugs, efforts have been done by scientists in various fields. In particular, Schiff bases play an essential role in biological fields as they are broadly used in various chemical, biological, and photochemical reactions. In recent times, the Schiff bases coordinated with the metals have been studied extensively in bioinorganic chemistry. The Schiff bases metal complexes are of great importance as they act as potential drugs which possess a wide spectrum of potential activity such as antimicrobial [4, 5], anticancer [6, 7], anti-inflammatory [8], antiparasitic [9], and antiviral [10]. From the detailed literature survey on condensed heterocyclic ring systems, it is evident that indenopyrazoles have also played a significant role due to their wide biological activities, viz., analgesics, antitubercular, anti-inflammatory agents, etc. An huge literature revealed that organotin(IV) complexes appeared as biologically potent moieties having antitumor [11, 12], antitubercular [13,14,15], antifertility [16, 17], antimicrobial [18,19,20], antiviral [21], antinematicidal, antiinsecticidal [22], antilieshmanial [23], anti-inflammatory [24], antidiabetic [25], antihypertensive [26], antioxidant [27], antimalarial [28] activity, etc. The bioactive metal complexes have enormous chemotherapeutic importance due to their ability to bind and degrade DNA of the microorganisms [29,30,31,32,33]. By considering the above facts and in continuation of our work towards the synthesis of bioactive compounds, we report herein the convenient synthesis, characterization, biological evaluation, and QSAR studies of Schiff bases containing oxygen and nitrogen donor atoms and their organotin (IV) complexes.
Results and discussion
The prior knowledge available for condensed heterocyclic ring systems indicated that indenopyrazoles have attained significant attention because of their synthetic utility and enormous biological potential. Indenopyrazoles have been found to be potent analgesics, antitubercular and anti-inflammatory agents, etc. Indenopyrazoles consist of indanedione moiety which may be responsible for their biological activities [34, 35]. The Schiff bases have also been reported to be good complexing agents. The Schiff base scaffolds play a vital role in the synthetic and medicinal chemistry as they are the core structure of numerous bioactive compounds. Hence, these types of moieties are getting attraction due to immense clinical importance [36, 37]. Therefore, based on the facts discussed above and continuing our research work, the synthesis, spectral characterization, in vitro antimicrobial potential and QSAR studies of diorganotin(IV) complexes of Schiff bases derived from, 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione with 4-substituted anilines are discussed in this section.
Chemistry
The starting triketone required for the synthesis of Schiff bases, i.e., 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione is synthesized by Claisen condensation of diethyl phthlate and 4-methylpentan-2-one in the presence of sodium methoxide as described in the literature [38, 39] (Scheme 1). The purity of the compound was confirmed by TLC and its melting point.

Further, the triketone 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione was condensed with 4-substituted anilines (p-methoxyaniline, p-nitroaniline, p-methylaniline, and p-chloroaniline) in methanol in an equimolar ratio which gave the desired Schiff bases in excellent yield. The condensation took place on side chain as reported in the literature. These Schiff bases were subsequently treated with dialkyl/aryltin(IV) dichloride to give respective diorganotin(IV) complexes in good yields. The synthetic protocol of Schiff bases derived from 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione with 4-substituted anilines and their diorganotin(IV) complexes is given in Scheme 2. All the diorganotin(IV) complexes were colored solids, stable, and soluble in CDCl3, DMSO, and MeOH. The Schiff base ligands and their diorganotin(IV) complexes were well characterized by elemental analysis, molar conductance, 1H, 13C, 119Sn NMR, and mass spectral studies. The ligands behaved as monobasic bidentate (O–/N–) and coordinated to the central tin atom with the oxygen atom of enol form. The molar conductance of these complexes was found in the range 10–17 Ω−1 cm2 mol−1 in DMSO, signifying the nonelectrolytic nature of complexes. The derived compounds were subjected to evaluation of their in vitro antimicrobial potential by serial dilution method. Quantitative structure–activity relationship (QSAR) analysis was performed using the linear free energy relationship model (LFER).

FT-IR spectra
The FT-IR spectra of all the synthesized compounds were recorded using KBr. The IR spectral data are given in the experimental section. The IR spectra of the Schiff bases displayed characteristic sharp absorption bands at 1720–1710 and 1597–1587 cm−1 due to C=O and C=N, respectively. By comparing the IR spectra of ligands and complexes, the coordination sites of the ligands were assigned. In the IR spectra of the complexes, absorption band due to C=N was shifted to lower frequency by 10–25 cm−1, which showed coordination through the azomethine nitrogen. The appearance of new band in the spectra of complexes at 1243–1069 cm−1 was due to C–O stretching confirmed that the coordination had occurred through the carbonyl oxygen to the tin after deprotonation. The new absorption bands in the complexes due to Sn–C, Sn–O, and Sn–N at 723–627, 553–531, and 475–451 cm−1, respectively, also confirmed the formation of the complexes [40, 41].
NMR spectra
The binding sites (O–/N–) of Schiff bases were further confirmed by comparing the 1H and 13C NMR spectra of ligands and complexes. The NMR spectra were recorded in CDCl3 and DMSO-d6. The spectral data are described in “Experimental” section.
1H NMR
In the 1H NMR spectra of the ligands, the signal due to enolic proton of the five membered ring of 1,3-indanedione moiety of Schiff base appeared as a singlet at δ = 12.47–12.28 ppm. The disappearance of this signal in the 1H NMR spectra of the complexes suggested the coordination mode of the carbonyl oxygen with central tin atom through enolization. In the 1H NMR spectra of ligands and complexes, isobutyl group exhibited a doublet in the region 3.41–2.96 ppm due to CH2 protons, a multiplet in the region 2.27–1.82 ppm due to CH proton, and a doublet for six protons of CH3 groups at 1.04–0.83 ppm. In the ligand H1L1 and its complexes, a singlet was observed due to three protons of OCH3 at 3.94–3.86 ppm due to electronegative oxygen atom, in ligand H1L3 and its complexes, three protons of CH3 appeared as a singlet in the region 2.32–2.30 ppm. The other aromatic protons appeared in the expected region. However, in the diorganotin(IV) complexes, some new signals appeared in the region 8.08–6.93 ppm due to phenyl group, 2.78–0.74 ppm due to butyl group, 1.75–0.83 ppm due to ethyl group, and 1.71–1.66 ppm due to methyl groups as expected [42].
13C NMR
The 13C NMR spectral data of all the synthesized compounds were found to be in the expected regions and further supported the data obtained from IR and 1H NMR. The characteristic signals in ligands due to carbonyl carbons and azomethine carbon were appeared at δ = 194.01–170.63 ppm and 159.02–148.66 ppm, respectively. The signal due to the C–H carbon of the five membered ring of 1,3-indanedione moiety of Schiff bases appeared at 103.69–100.83 ppm for ligands and complexes supporting the tautomerization of the proton attached to the carbon. In 13C NMR spectra of ligands and complexes signals due to carbons of OCH3 and CH3 were observed at 57.35–55.54 ppm and 39.63–34.72 ppm, respectively. The three signals of isobutyl moiety appeared in the range 38.96–35.45, 29.09–22.96, and 22.69–21.23 ppm can be assigned to CH2, CH, CH3 carbons, respectively. The signals due to carbonyl carbon, azomethine carbon and the carbon directly attached to the central metal were shifted downfield on complexation owing to decrease in electron density on these groups and electropositive nature of tin metal. The data supported the coordination modes through azomethine nitrogen and carbonyl carbon. In the 13C spectra of the complexes, the new signals due to carbon of methyl, ethyl, butyl, and phenyl group directly attached to central tin atom appeared in the expected region [43].
119Sn NMR
The pentacoordinated environment of the diorganotin(IV) complexes was confirmed by 119Sn NMR spectra. In 119Sn spectra of complexes, a sharp singlet was observed indicating the formation of a single tin species. In 119Sn NMR spectra signal in the range δ = − 397.48 to − 378.32 ppm, − 298.21 to − 273.18 ppm, − 233.60 to − 203.17 ppm, and − 189.66 to − 178.30 ppm was due to phenyl, butyl, ethyl, and methyl complexes which were in accordance with the literature and supported the pentacoordinated environment and distorted trigonal bipyramidal geometry around the tin atom [44, 45].
Mass spectra
The ESI-mass spectra of all the synthesized compounds further confirmed the formation of the compounds as the data were found to be in good agreement with their molecular formula (vide experimental). The fragmentation patterns were analysed and base peaks were found due to [L]+ and the peak due to [M+H]+ ion of the diorganotin(IV) complexes was found with very low abundance in some cases. In the mass spectra of the complexes, the fragment ions were observed due to the [M+H]+, [R′2SnL]+, [L]+, [R′SnL]+, [SnL]+, [Sn]+ which were in accordance with literature [46]. The molecular ion and base peaks of Ph2SnClL1 were appeared at m/z = 644.50 and 336.00 due to [M+H]+ and [L]+, respectively, which were found in agreement with the theoretical values. The ESI-mass spectrum of diorganotin(IV) complex 1 is shown in Fig. 1.

ESI-mass spectrum of diorganotin(IV) complex 1
In vitro antimicrobial activity
The Schiff bases derived from 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione with 4-substituted anilines and their diorganotin(IV) complexes were tested for their potential against Gram-positive bacterial strains, viz., Staphylococcus aureus, Bacillus cereus, Gram-negative bacterial strains, viz., Escherichia coli, Pseudomonas aeruginosa, and fungal strains, viz., Aspergillus flavus, Aspergillus niger, and Candida albicans using serial dilution technique and ciprofloxacin and fluconazole as standard drugs. The potential regarding the antibacterial and antifungal activity is presented in Table 1 and Fig. 2. The presence of C=N group in the Schiff bases and central tin atom plays a vital role in biological activity. Among the tested compounds, most of the complexes were found more potent against microbial strains as the R′, group increases the lipophilicity of the complexes as they can easily bind with biological molecules by π–π interactions which boost up their bioactivity. The diorganotin(IV) complexes were found to be more potent as compared to the Schiff base ligands against the same strains as explained by the chelation theory [47], which is responsible for the augmentation of the biocidal activity. The compounds penetrate through the cell wall and bind with the DNA of the microorganism which leads to deactivate the respiration process of the microorganisms. The hydrogen bond formation between oxygen and nitrogen atom with active centers of cell constituents may alter the mode of action at cell processes [48, 49]. Further, the derived compounds had higher potential against Gram-positive bacterial strains as compared to Gram negative due to the presence of outer complex cell membrane, containing lipopolysaccharides. The data revealed that the complexes 13 (MIC 0.0048 µmol/cm3) against E. coli and 1 (MIC 0.0024 µmol/cm3) against P. aeruginosa, 5 (MIC 0.0047 µmol/cm3) against B. cereus, 1 (MIC 0.0024 µmol/cm3) against S. aureus, 1 (MIC 0.0024 µmol/cm3) against A. flavus, 5 (MIC 0.0024 µmol/cm3) against A. niger and 1 (MIC 0.0024 µmol/cm3) against C. albicans (MIC 0.0024 µmol/cm3) were found to be most potent.

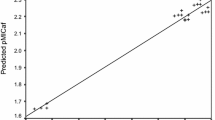
Plot of observed pMICpa against the predicted pMICpa for the QSAR model developed by Eq. (1)
QSAR studies
In the present study, data set of twenty compounds (H1L1–H1L4 and diorganotin(IV) complexes 1–16) was submitted for the regression analysis to correlate the molecular descriptors of these compounds with the observed in vitro antimicrobial activities. During the regression analysis study, it was observed that the results of the two compounds, viz., 1 and 16 were outside the limits of whole data set and being outliers were not included in the regression analysis for the generation of mathematical models for the prediction of antimicrobial activities. The relationship between molecular descriptors and biological potential was accessed by regression analysis and correlation matrix constructed for antibacterial activity against P. aeruginosa is presented in Table 2 (supplementary section). The interrelationship of various structural descriptors with antimicrobial potential is presented in Table 3 (supplementary section). In general, going through the whole data set, high colinearity (r > 0.8) was observed between different parameters, i.e., molecular descriptors. The active interrelationship was observed between zero-order molecular connectivity index, 0χ and electronic energy (r = 0.999), nuclear repulsion energy, and zero-order molecular connectivity index, 0χ (r = 0.998), and least interrelationship was found between Balaban index (J), and dipole moment, μ (r = 0.007) and third-order molecular connectivity index, 3χ and energy of highest occupied molecular orbital, HOMO (r = 0.009). The correlation matrix pointed the role of kappa shape indices, Randic parameter, and molecular connectivity indices in the expression of antibacterial and antifungal activity by the synthesized complexes [50, 51].
The assessment of antimicrobial potential of synthesized compounds against P. aeruginosa revealed that topological descriptor first-order kappa shape index (κ1) was effectively controlling the antibacterial activity (Eq. 1).
QSAR model for antibacterial activity against P. aeruginosa is
where r is the correlation coefficient, n is the number of data points, r2 is the squared correlation coefficient, q2 is the cross-validated r2 obtained by leave one out method, s is the standard error of the estimate and F is the Fischer statistics.
The QSAR model for antibacterial activity against P. aeruginosa (Eq. 1) demonstrated the role of first-order Kappa shape indices (κ1) in modulating the activity. According to Kier, “the shape of a molecule may be partitioned into attributes, each described by the count of bonds of various path lengths. The basis for devising a relative index of shape is given by the relationship of the number of path of length l in the molecule i, lPi, to some reference values based on molecules with a given number of atoms, n, in which the values of lP are maximum and minimum, lPmax and lPmin. The first-order shape attribute, κ1, is given by the following expression”:
It can be seen from the results of antimicrobial activity in Table 4 (supplementary section) that compounds 1 and 5 have the highest antibacterial potential against P. aeruginosa and they have high κ1 values (Table 5, supplementary section) which are in concordance with the model expressed by Eq. (1) in which there is a positive correlation between antibacterial potential of synthesized compounds and topological descriptors κ1. The best use of QSAR models is in their ability to predict the activity potential of similar compounds and this assessing potential is measured by testing the validity of derived QSAR models. The validity of the models was measured by the leave one out (LOO) method and q2 value was found to be 0.910. The QSAR model is valid if value of q2 > 0.5; hence, the model represented by Eq. (1) was considered as a valid one. Further, the mathematical model (Eq. 1) was also tested for its predictability and the results of predicted antibacterial activity are presented in Table 6 (supplementary section).
The comparison of in vitro observed and predicted antibacterial activity indicated that the model represented by Eq. (1) was valid as it gave low residual values, hence supporting the validity of the derived model. Further, to confirm the predictability and validity of derived QSAR model a plot of observed and predicted antibacterial activity against P. aeruginosa was drawn which also supported the validity of model represented by Eq. (1) (Fig. 2). The plot of observed antibacterial activity against residual antibacterial activity (pMICpa) demonstrated the nonexistence of systematic errors in development of QSAR model (Fig. 3) [52].

Plot of residual pMICpa against the observed pMICpa for the QSAR model developed by Eq. (1)
The other important QSAR models derived are as represented below.
QSAR model for antibacterial activity against E. coli is
QSAR model for antibacterial activity against E. coli is
QSAR model for antifungal activity against A. niger is
QSAR model for antibacterial activity against B. cereus is
QSAR model for antibacterial activity against S. aureus is
QSAR model for antifungal activity against C. albicans is
The above models have been discussed in detail in the supplementary section.
Conclusion
Diorganotin(IV) complexes were obtained by the reaction of 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione and 4-substituted anilines (p-OCH3, p-NO2, p-CH3, p-Cl) with R2SnCl2, (R=Me, Et, n-Bu, Ph). The synthesized complexes have been characterized by different spectroscopic (1H, 13C, 119Sn NMR, IR, mass) and other physical techniques. The Schiff base ligands were found to be bidentate coordinated with tin metal with (N–/O–) donor sites having pentacoordinated tin(IV) complexes. These compounds were further screened for their in vitro antimicrobial activity against different bacterial and fungal strains. The activity varies with the substitution on the tin atom, and the phenyl complexes were found to be more potent than other compounds. The presence of chlorine atom showed superior biocidal activity. Furthermore, the QSAR analysis revealed that the antimicrobial activity was controlled by topological indices which indicated that the complexes were more prolific than the parent ligands.
Experimental
All the reactions were carried under inert atmosphere. The starting materials, i.e., dimethyltin(IV) dichloride, diethyltin(IV) dichloride, di-n-butyltin(IV) dichloride, and diphenyltin(IV) dichloride were of analytical grade supplied by Sigma-Aldrich and used as such without any further purification. The elemental analysis (C, H, and N) was carried on a Perkin-Elmer 2400 instrument and the measured data were found to be in accordance with the calculated data. The Fourier transform infrared (FTIR) spectra (4000–400 cm−1) were recorded on a Perkin-Elmer spectrum RX1 instrument. The NMR spectra were recorded on a Bruker Avance II 400 MHz NMR spectrometer in CDCl3 or DMSO-d6 using tetramethylsilane (TMS) as an internal standard. The mass spectra were recorded on an LCMS MS 6410 Agilent Technologies spectrometer with an electron impact quadropole analyzer. Tin was estimated gravimetrically as SnO2.
General procedure for the synthesis of Schiff base ligands H 1 L 1–H 1 L 4
The 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione needed for the synthesis was prepared by the Claisen condensation of diethyl phthlate and 4-methylpentan-2-one in the presence of sodium methoxide as described in the literature. procedure [38, 39]. Schiff base ligands H1L1–H1L4 were prepared in good yield by the reaction of 1.151 g 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione (5 mmol) and 0.615 g p-methoxyaniline (5 mmol) or 0.690 g p-nitroaniline (5 mmol) or 0.535 g p-methylaniline (5 mmol) or 0.637 g p-chloroaniline (5 mmol) in 1:1 molar ratio in 30 cm3 methanol by adding 3–4 drops of glacial acetic acid. The progress of reaction was monitored by TLC analysis of the reaction mixture withdrawn at different interval of time. The reaction mixture was refluxed for 6–7 h and after the completion of reaction, contents were allowed to cool and stand overnight at room temperature. The products thus obtained were filtered, washed with methanol, and recrystallized from methanol and chloroform solution to furnish the pure products.
(E)-2-[1-(4-Methoxyphenylimino)-3-methylbutyl]-1H-indene-1,3(2H)-dione (H 1 L 1, C21H21NO)
White solid; yield: 93%; m.p.: 174–176 °C; IR (KBr): \( \bar{\nu } \) = 1716 (C=O), 1587 (C=N) cm−1; 1H NMR (CDCl3): δ = 12.28 (s, 1H), 7.72 (d, 2H), 7.59 (d, 2H, J = 8 Hz), 7.14 (d, 2H, J = 8 Hz), 6.95 (d, 2H, J = 8 Hz), 3.86 (s, 3H), 2.97 (d, 2H, J = 8 Hz), 2.00–1.90 (m, 1H), 0.86 (d, 6H, J = 4 Hz) ppm; 13C NMR (CDCl3): δ = 195.0, 190.4, 166.0, 158.9, 139.9, 138.9, 133.2, 132.8, 129.1, 127.7, 121.4, 120.9, 114.6, 103.6, 55.5, 35.5, 29.0, 22.4 ppm; ESI–MS: m/z calcd. for C21H21NO ([M+H]+) 336.16, found 336.10.
(E)-2-[3-Methyl-1-(4-nitrophenylimino)butyl]-1H-indene-1,3(2H)-dione (H 1 L 2, C20H18N2O4)
White solid; yield: 89%; m.p.: 178–181 °C; IR (KBr): \( \bar{\nu } \) = 1720 (C=O), 1590 (C=N) cm−1; 1H NMR (CDCl3): δ = 12.61 (s, 1H), 8.07 (d, 2H), 7.84 (d, 2H, J = 8 Hz), 7.41 (d, 2H, J = 8 Hz), 6.63 (d, 2H, J = 8 Hz), 2.86 (d, 2H, J = 8 Hz), 2.27–2.18 (m, 1H), 1.04 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 191.4, 189.4, 157.7, 148.9, 142.6, 134.6, 132.3, 126.3, 124.5, 101.5, 38.9, 24.4, 22.7 ppm; ESI–MS: m/z calcd. for C20H18N2O4 ([M+H]+) 351.3, found 351.60.
(E)-2-[3-Methyl-1-(p-tolylimino)butyl]-1H-indene-1,3(2H)-dione (H 1 L 3, C21H21NO2)
White solid; yield: 87%; m.p.: 201–203 °C; IR (KBr): \( \bar{\nu } \) = 1718 (C=O), 1595 (C=N) cm−1; 1H NMR (CDCl3): δ = 12.47 (s, 1H), 7.87 (d, 2H), 7.62 (d, 2H, J = 8 Hz), 7.33 (d, 2H, J = 8 Hz), 7.15 (d, 2H, J = 8 Hz), 2.31 (s, 3H), 2.96 (d, 2H, J = 8 Hz), 1.93–1.82 (m, 1H), 0.91 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 193.7, 188.3 (C=O), 153.6 (C=N), 140.2, 134.5, 132.4, 128.6, 127.8, 126.7, 100.8, 39.6, 30.7, 22.9, 21.5 ppm; ESI–MS: m/z calcd. for C21H21NO2 ([M+H]+) 320.17, found 320.20.
(E)-2-[1-(4-Chlorophenylimino)-3-methylbutyl]-1H-indene-1,3(2H)-dione (H 1 L 4, C20H18ClNO2)
White solid; yield: 91%; m.p.: 191–193 °C; IR (KBr): \( \bar{\nu } \) = 1710 (C=O), 1597 (C=N) cm−1; 1H NMR (CDCl3): δ = 12.31 (s, 1H), 7.73 (m, 2H), 7.61 (m, 2H, J = 8 Hz), 7.23–7.13 (m, 4H), 2.97 (d, 2H, J = 8 Hz), 1.98–1.89 (m, 1H), 0.86 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 192.6, 189.6, 156.7 (C=N), 145.5, 132.8, 130.3, 129.7, 128.8, 127.4, 101.3, 35.5, 23.6, 22.4 ppm; ESI–MS: m/z calcd. for C20H18ClNO2 ([M+H]+) 340.11, found 340.20.
General procedure for the synthesis of diorganotin(IV) complexes 1–16
For the synthesis of diorganotin(IV) complexes firstly sodium salt of the ligands by the reaction of 1.67 g H1L1, 1.75 g H1L2, 1.59 g H1L3, or 1.69 g H1L4 (5 mmol each) and 0.23 g sodium metal (5 mmol) in 20 cm3 dry methanol was prepared. To the reaction mixture, the methanolic solution of 1.71 g diphenyltin(IV) dichloride, 1.51 g dibutyltin(IV) dichloride, 1.23 g diethyltin(IV) dichloride, or 1.09 g dimethyltin(IV) dichloride was added dropwise with constant stirring and then refluxed at 40–50 °C for 6–7 h. Thereafter, the reaction mixture was cooled and kept overnight at room temperature. Then, the reaction mixture was filtered to remove sodium chloride salt thus formed. Consequently, the excess solvent was evaporated over rotary evaporator under reduced pressure to separate out the solid. The solid thus obtained was washed with dry hexane and recrystallized with methanol and chloroform mixture to get the pure products.
(E)-3-[(Chlorodiphenylstannyl)oxy]-2-[1-(4-methoxyphenylimino)-3-methylbutyl]-1H-inden-1-one (1, C33H30ClNO3Sn)
Yellow solid; yield: 76%; m.p.: 187–189 °C; IR (KBr): \( \bar{\nu } \) = 1708 (C=O), 1568 (C=N), 1172 (C–O), 692 (Sn–C), 541 (Sn–O), 471 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.90–6.99 (m, 18H), 3.87 (s, 3H), 2.97 (d, 2H, J = 8 Hz), 1.98–1.91 (m, 1H), 0.86 (d, 6H) ppm; 13C NMR (CDCl3): δ = 195.0, 190.4, 169.0, 158.0, 139.8, 138.9, 136.1, 133.2, 132.8, 129.1, 127.7, 121.4, 121.0, 114.6, 103.6, 55.6, 35.4, 29.0, 22.4 ppm; 119Sn NMR (CDCl3): δ = − 379.47 ppm; ESI–MS: m/z calcd. for C33H30ClNO3Sn ([M+H]+) 644.10, found 644.40.
(E)-3-[(Dibutylchlorostannyl)oxy]-2-[1-(4-methoxyphenylimino)-3-methylbutyl]-1H-inden-1-one (2, C29H38ClNO3Sn)
Yellow solid; yield: 78%; m.p.: 182–183 °C; IR (KBr): \( \bar{\nu } \) = 1718 (C=O), 1571 (C=N), 1109 (C–O), 627 (Sn–C), 537 (Sn–O), 462 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.57 (d, 2H), 7.31 (d, 2H, J = 8 Hz), 7.09 (d, 2H, J = 8 Hz), 6.92 (d, 2H, J = 8 Hz), 3.91 (s, 3H), 3.41 (d, 2H, J = 8 Hz), 2.78–2.47 (m, 6H), 2.11–2.03 (m, 1H, CH), 1.48–0.83 (m, 18H) ppm; 13C NMR (CDCl3): δ = 194.0, 189.6, 168.6, 157.7, 139.4, 137.1, 135.6, 133.3, 131.3, 129.2, 101.0, 56.5, 35.4, 30.1, 26.7, 25.5, 23.1, 22.7, 15.9 ppm; 119Sn NMR (CDCl3): δ = -287.19 ppm; ESI–MS: m/z calcd. for C29H38ClNO3Sn ([M+H]+) 604.16, found 604.40.
(E)-3-[(Diethylchlorostannyl)oxy]-2-[1-(4-methoxyphenylimino)-3-methylbutyl]-1H-inden-1-one (3, C25H30ClNO3Sn)
Yellow solid; yield: 72%; m.p.: 189–191 °C; IR (KBr): \( \bar{\nu } \) = 1709 (C=O), 1573 (C=N), 1075 (C–O), 671 (Sn–C), 539 (Sn–O), 473 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.79 (d, 2H, J = 8 Hz), 7.71–7.63 (m, 2H), 7.19 (d, 2H), 7.05 (d, 2H, J = 8 Hz), 3.93 (s, 3H), 2.94 (d, 2H, J = 8 Hz), 2.13–2.02 (m, 1H), 1.73–1.63 (m, 4H), 1.21–0.83 (m, 12H) ppm; 13C NMR (CDCl3): δ = 193.1, 189.0, 168.8, 158.6, 141.0, 137.7, 133.5, 131.4, 130.2, 129.7, 102.8, 56.7, 36.1, 29.6, 25.3, 21.5, 15.2 ppm; 119Sn NMR (CDCl3): δ = − 203.17 ppm; ESI–MS: m/z calcd. for C25H30ClNO3Sn ([M+H]+) 548.10, found 548.90.
(E)-3-[(Chlorodimethylstannyl)oxy]-2-[1-(4-methoxyphenylimino)-3-methylbutyl]-1H-inden-1-one (4, C23H26ClNO3Sn)
Yellow solid; yield: 78%; m.p.: 185–187 °C; IR (KBr): \( \bar{\nu } \) = 1721 (C=O), 1572 (C=N), 1069 (C–O), 679 (Sn–C), 538 (Sn–O), 469 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.80 (m, 2H), 7.70–7.65 (m, 2H), 7.22 (d, 2H, J = 8 Hz), 7.03 (d, 2H, J = 8 Hz), 3.94 (s, 3H), 3.05 (d, 2H, J = 8 Hz), 2.07–2.00 (m, 1H), 1.66 (s, 6H), 0.94 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 194.39, 190.0, 171.7, 157.0, 140.8, 138.3, 134.4, 132.1, 131.7, 129.1, 99.2, 57.3, 35.5, 26.7, 21.2, 15.2 ppm; 119Sn NMR (CDCl3): δ = − 189.66 ppm; ESI–MS: m/z calcd. for C23H26ClNO3Sn ([M+H]+) 520.07, found 520.60.
(E)-3-[(Chlorodiphenylstannyl)oxy]-2-[3-methyl-1-(4-nitrophenylimino)butyl]-1H-inden-1-one (5, C32H27ClN2O4Sn)
Yellow solid; yield: 90%; m.p.: 172 °C; IR (KBr): \( \bar{\nu } \) = 1716 (C=O), 1577 (C=N), 1191 (C–O), 701 (Sn–C), 546 (Sn–O), 474 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 8.08 (d, 2H, J = 8 Hz), 7.81–6.93 (m, 16H), 2.96 (d, 2H, J = 8 Hz), 1.92–1.87 (m, 1H), 0.83 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 193.5, 189.0, 162.9, 147.2, 139.4, 138.3, 137.8, 136.2, 133.0, 132.2, 129.1, 127.9, 122.6, 116.7, 101.0, 35.7, 27.8, 22.1 ppm; 119Sn NMR (CDCl3): δ = − 393.89 ppm; ESI–MS: m/z calcd. for C32H27ClN2O4Sn ([M+H]+) 659.08, found 659.40.
(E)-3-[(Dibutylchlorostannyl)oxy]-2-[3-methyl-1-(4-nitrophenylimino)butyl]-1H-inden-1-one (6, C28H35ClN2O4Sn)
Yellow solid; yield: 82%; m.p.: 198–200 °C; IR (KBr): \( \bar{\nu } \) = 1719 (C=O), 1572 (C=N), 1088 (C–O), 665 (Sn–C), 544 (Sn–O), 468 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 8.06 (d, 2H, J = 8 Hz), 7.87 (d, 2H, J = 8 Hz), 7.42 (d, 2H, J = 8 Hz), 6.73 (d, 2H, J = 8 Hz), 3.31 (d, 2H, J = 8 Hz), 2.51–2.48 (m, 3H), 1.52–0.84 (m, 21H) ppm; 13C NMR (CDCl3): δ = 192.8, 188.2, 159.0, 146.9, 139.1, 137.4, 135.6, 133.4, 132.4, 129.0, 100.0, 36.5, 28.1, 25.2, 22.1, 21.7, 20.1, 13.3 ppm; 119Sn NMR (CDCl3): δ = − 273.18 ppm; ESI–MS: m/z calcd. for C28H35ClN2O4Sn ([M+H]+) 619.14, found 619.60.
(E)-3-[(Chlorodiethylstannyl)oxy]-2-[3-methyl-1-(4-nitrophenylimino)butyl]-1H-inden-1-one (7, C24H27ClN2O4Sn)
Yellow solid; yield: 90%; m.p.: 172 °C; IR (KBr): \( \bar{\nu } \) = 1714 (C=O), 1568 (C=N), 1092 (C–O), 688 (Sn–C), 531 (Sn–O), 460 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 8.07 (d, 2H, J = 8 Hz), 7.79 (d, 2H, J = 8 Hz), 7.41 (d, 2H), 6.93 (d, 2H, J = 8 Hz), 2.99 (d, 2H, J = 8 Hz), 2.07–1.98 (m, 1H), 1.75–1.68 (m, 4H), 1.23–0.88 (m, 12H) ppm; 13C NMR (CDCl3): δ = 194.4, 190.6, 158.6, 147.5, 141.9, 139.2, 137.1, 133.3, 131.2, 128.4, 100.2, 35.7, 28.8, 26.1, 21.7, 15.5 ppm; 119Sn NMR (CDCl3): δ = − 213.70 ppm; ESI–MS: m/z calcd. for C24H27ClN2O4Sn ([M+H]+) 563.08, found 563.10.
(E)-3-[(Chlorodimethylstannyl)oxy]-2-[3-methyl-1-(4-nitrophenylimino)butyl]-1H-inden-1-one (8, C22H23ClN2O4Sn)
Yellow solid; yield: 81%; m.p.: 191–193 °C; IR (KBr): \( \bar{\nu } \) = 1711(C=O), 1573 (C=N), 1093 (C–O), 657 (Sn–C), 538 (Sn–O), 475 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 8.03 (d, 2H, J = 8 Hz), 7.86 (d, 2H, J = 8 Hz), 7.44 (d, 2H, J = 8 Hz), 6.83 (d, 2H, J = 8 Hz), 3.01 (d, 2H, J = 8 Hz), 2.08–1.99 (m, 1H), 1.68 (s, 6H), 0.93 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 193.4, 189.7, 158.4, 146.0, 140.8, 139.2, 138.6, 134.4, 133.4, 129.5, 99.9, 36.2, 29.7, 23.3, 14.5 ppm; 119Sn NMR (CDCl3): δ = − 182.36 ppm; ESI–MS: m/z calcd. for C22H23ClN2O4Sn ([M+H]+) 535.04, found 535.60.
(E)-3-[(Chlorodiphenylstannyl)oxy]-2-[3-methyl-1-(p-tolylimino)butyl]-1H-inden-1-one (9, C33H30ClNO2Sn)
Yellow solid; yield: 90%; m.p.: 197–199 °C, IR (KBr): \( \bar{\nu } \) = 1721 (C=O), 1582 (C=N), 1171 (C–O), 723 (Sn–C), 542 (Sn–O), 461 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.79–6.97 (m, 18H), 2.94 (d, 2H, J = 8 Hz), 2.32 (s, 3H), 1.90–1.79 (m, 1H), 0.89 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 194.7, 189.3, 162.0, 142.9, 137.5, 136.4, 133.1, 132.2, 130.9, 129.7, 128.3, 127.3, 122.1, 115.47, 100.6, 36.9, 31.1, 22.3, 21.2 ppm; 119Sn NMR (CDCl3): δ = − 397.48 ppm; ESI–MS: m/z calcd. for C33H30ClNO2Sn ([M+H]+) 628.11, found 628.40.
(E)-3-[(Dibutylchlorostannyl)oxy]-2-[3-methyl-1-(p-tolylimino)butyl]-1H-inden-1-one (10, C29H38ClNO2Sn)
Yellow solid; yield: 89%; m.p.: 210–212 °C; IR (KBr): \( \bar{\nu } \) = 1699 (C=O), 1578 (C=N), 1243 (C–O), 705 (Sn–C), 553 (Sn–O), 467 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.73 (d, 2H, J = 8 Hz), 7.64 (d, 2H, J = 8 Hz), 7.31 (d, 2H, J = 8 Hz), 7.18 (d, 2H, J = 8 Hz), 3.37 (d, 2H, J = 8 Hz), 2.48–2.38 (m, 5H), 2.31 (s, 3H), 2.11–2.01 (m, 1H), 1.42–0.81 (m, 19H) ppm; 13C NMR (CDCl3): δ = 194.3, 188.7, 159.4, 140.0, 138.2, 135.5, 133.6, 131.9, 130.5, 128.9, 100.0, 38.7, 30.2, 27.1, 25.7, 23.6, 22.5, 21.1, 14.1 ppm; 119Sn NMR (CDCl3): δ = − 298.21 ppm; ESI–MS: m/z calcd. for C29H38ClNO2Sn ([M+H]+) 588.17, found 588.40.
(E)-3-[(Chlorodiethylstannyl)oxy]-2-[3-methyl-1-(p-tolylimino)butyl]-1H-inden-1-one (11, C25H30ClNO2Sn)
Yellow solid; yield: 82%; m.p.: 198–200 °C; IR (KBr): \( \bar{\nu } \) = 1712 (C=O), 1587 (C=N), 1108 (C–O), 692 (Sn–C), 548 (Sn–O), 471 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.78 (d, 2H, J = 8 Hz), 7.66 (d, 2H, J = 8 Hz), 7.33 (d, 2H), 7.24 (d, 2H, J = 8 Hz), 2.99 (d, 2H, J = 8 Hz), 2.32 (s, 3H), 2.15–2.04 (m, 1H), 1.77–1.69 (m, 4H), 1.32–0.91 (m, 12H) ppm; 13C NMR (CDCl3): δ = 193.4, 189.3, 158.6, 141.2, 138.2, 136.7, 133.3, 130.1, 126.7, 129.4, 100.2, 35.3, 27.6, 25.4, 22.1, 21.5, 15.5 ppm; 119Sn NMR (CDCl3): δ = − 233.60 ppm; ESI–MS: m/z calcd. for C25H30ClNO2Sn ([M+H]+) 532.11, found 532.70.
(E)-3-[(Chlorodimethylstannyl)oxy]-2-[3-methyl-1-(p-tolylimino)butyl]-1H-inden-1-one (12, C23H26ClNO2Sn)
Yellow solid; yield: 90%; m.p.: 201–203 °C; IR (KBr): \( \bar{\nu } \) = 1709 (C=O), 1588 (C=N), 1073 (C–O), 689 (Sn–C), 543 (Sn–O), 463 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.79 (d, 2H, J = 8 Hz), 7.68 (d, 2H, J = 8 Hz), 7.34 (d, 2H, J = 8 Hz), 7.21 (d, 2H, J = 8 Hz), 2.98 (d, 2H, J = 8 Hz), 2.30 (s, 3H), 2.11–2.01 (m, 1H), 1.69 (s, 3H), 0.87 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 195.1, 189.8, 159.4, 140.8, 139.2, 138.6, 136.7, 133.4, 130.4, 128.7, 100.9, 34.7, 28.8, 24.3, 21.6, 15.0 ppm; 119Sn NMR (CDCl3): δ = − 183.06 ppm; ESI–MS: m/z calcd. for C23H26ClNO2Sn ([M+H]+) 504.08, found 504.90.
(E)-3-[(Chlorodiphenylstannyl)oxy]-2-[1-(4-chlorophenylimino)-3-methylbutyl]-1H-inden-1-one (13, C32H27Cl2NO2Sn)
Yellow solid; yield: 85%; m.p.: 232–234 °C; IR (KBr): \( \bar{\nu } \) = 1722 (C=O), 1577 (C=N), 1196 (C–O), 683 (Sn–C), 535 (Sn–O), 458 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.63–7.12 (m, 18H), 2.96 (d, 2H, J = 8 Hz), 1.91–1.82 (m, 1H), 0.81 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 193.8, 189.0, 158.5, 144.0, 139.9, 136.1, 133.7, 133.2, 129.3, 128.9, 128.5, 125.6, 122.4, 117.4, 100.9, 35.7, 29.2, 22.3 ppm; 119Sn NMR (CDCl3): δ = − 391.47 ppm; ESI–MS: m/z calcd. for C32H27Cl2NO2Sn ([M+H]+) 648.05, found 648.40.
(E)-2-[1-(4-Chlorophenylimino)-3-methylbutyl]-3-[(dibutylchlorostannyl)oxy]-1H-inden-1-one (14, C28H35Cl2NO2Sn)
Yellow solid; yield: 76%; m.p.: 211–213 °C; IR (KBr): \( \bar{\nu } \) = 1718 (C=O), 1568 (C=N), 1201 (C–O), 678 (Sn–C), 543 (Sn–O), 472 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.64–7.42 (m, 5H), 7.31–7.13 (m, 2H), 6.88–6.78 (m, 1H), 3.19–2.94 (m, 7H), 2.71 (d, 2H, J = 8 Hz), 2.13–2.04 (m, 1H), 1.82–0.74 (m, 17H) ppm; 13C NMR (CDCl3): δ = 193.7, 188.3, 159.6, 145.0, 137.2, 136.5, 133.2, 132.1, 130.9, 129.4, 98.0, 37.7, 28.7, 26.8, 25.4, 21.5, 20.4, 13.8 ppm; 119Sn NMR (CDCl3): δ = − 273.46; ESI–MS: m/z calcd. for C28H35Cl2NO2Sn ([M+H]+) 608.11, found 608.60.
(E)-3-[(Chlorodiethylstannyl)oxy]-2-[1-(4-chlorophenylimino)-3-methylbutyl]-1H-inden-1-one (15, C24H27Cl2NO2Sn)
Yellow solid; yield: 86%; m.p.: 202–204 °C; IR (KBr): \( \bar{\nu } \) = 1720 (C=O), 1567 (C=N), 1166 (C–O), 702 (Sn–C), 543 (Sn–O), 461 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.61–7.39 (m, 4H), 7.22 (d, 2H, J = 8 Hz), 6.90 (d, 2H, J = 8 Hz), 2.97 (d, 2H, J = 8 Hz), 2.27–2.19 (d, 2H), 2.09–1.74 (m, 1H), 1.64–1.52 (m, 4H), 1.30–0.90 (m, 12H) ppm; 13C NMR (CDCl3): δ = 192.4, 187.5, 157.1, 144.0, 139.2, 138.5, 134.1, 133.4, 129.3, 125.7, 100.8, 35.9, 29.6, 25.2, 21.6, 15.4 ppm; 119Sn NMR (CDCl3): δ = − 203.38 ppm; ESI–MS: m/z calcd. for C24H27Cl2NO2Sn ([M+H]+) 552.05, found 552.70.
(E)-3-[(Chlorodimethylstannyl)oxy]-2-[1-(4-chlorophenylimino)-3-methylbutyl]-1H-inden-1-one (16, C22H23Cl2NO2Sn)
Yellow solid; yield: 82%; m.p.: 189–191 °C; IR (KBr): \( \bar{\nu } \) = 1709 (C=O), 1562 (C=N), 1189 (C–O), 697 (Sn–C), 552 (Sn–O), 451 (Sn–N) cm−1; 1H NMR (CDCl3): δ = 7.62–7.38 (m, 4H), 7.22 (d, 2H, J = 8 Hz), 6.89 (d, 2H, J = 8 Hz), 2.96 (d, 2H, J = 8 Hz), 2.01–1.91 (m, 1H), 1.71 (s, 6H), 0.93 (d, 6H, J = 8 Hz) ppm; 13C NMR (CDCl3): δ = 194.3, 189.1, 159.9, 140.2, 139.0, 136.1, 133.6, 131.5, 128.2, 126.1, 102.1, 35.9, 25.1, 21.7, 16.3 ppm; 119Sn NMR (CDCl3): δ = − 178.30 ppm; ESI–MS: m/z calcd. for C22H23Cl2NO2Sn ([M+H]+) 524.02, found 524.70.
Pharmacology
All the newly synthesized compounds were assessed for their in vitro antimicrobial activity against Gram positive bacterial strains, viz., B. cereus (MTCC 10072), S. aureus (NCIM 2901) and Gram negative bacterial strains, viz., E. coli (MTCC 732), P. aeruginosa (MTCC 424), and fungal strains, viz., A. flavus (ATCC 76801), A. niger (MTCC 9933), and C. albicans (MTCC 227) at different concentrations under the standard set of conditions. Minimum inhibitory concentration (MIC, µmol/cm3) was calculated by serial dilution method.
QSAR studies of the Schiff bases and their diorganotin(IV) complexes
For QSAR study a data set of synthesized compounds with different antimicrobial activities excluding outliers were used. The antimicrobial activity was calculated in MIC (µmol/cm3) and it was converted into the corresponding pMIC (− logMIC). The structures of the molecules were optimized by using Marvin Sketch. The molecular parameters including one-, two-, and three-dimensional descriptors were calculated. The pre-optimization of the structures of test compound was performed by molecular mechanics force field (MM) process of Hyperchem 6.03 [53] and the resultant geometries were further developed by means of the semiempirical method PM3 (Parametric Method 3). A gradient norm limit of 0.01 kcal/Ǻ was used for the geometry optimization. The minimum energy structure of every molecule was taken for the calculation of physicochemical parameters using TSAR 3.3 software for Windows [54]. The regression analysis was done using the SPSS software package [55]. Predictive power and model’s corpulence were assessed by means of cross-validation coefficient q2.
References
Thumar NJ, Patel MP (2012) Med Chem Res 21:1751
Peto J (2011) Nature 411:390
Chopra I, Schofield C, Everett M, O’Neill A, Miller K, Wilcox M, Frere JM, Dawson M, Czapleavski L, Courvalin P (2008) Lancet Infect Dis 8:133
Zamudio-Rivera LS, George-Tellez R, López-Mendoza G, Morales-Pacheco A, Flores E, Höpfl H, Barba V, Fernandez FJ, Cabirol N, Beltrán HI (2005) Inorg Chem 44:5370
Gaur S, Fahmi N, Singh RV (2007) Phosphorus. Sulfur Silicon Relat Elem 182:853
Khan RA, Yadav S, Hussain Z, Arjmand F, Tabassum S (2014) Dalton Trans 43:2534
Shpakovsky DB, Banti CN, Mukhatova EM, Gracheva YA, Osipova VP, Berberova NT, Albov DV, Antonenko TA, Aslanov LA, Milaeva ER, Hadjikakou SK (2014) Dalton Trans 43:6880
Bekhit AA, Fahmy HT, Rostom SA, Baraka AM (2003) Eur J Med Chem 38:27
Rathelot P, Azas N, El-Kashef H, Delmas F, Di Giorgio C, Timon-David P, Maldonado J, Vanelle P (2002) Eur J Med Chem 37:671
Chavan SP, Sivappa R (2004) Tetrahedron Lett 45:3941
de Oliveira KN, Andermark V, Onambele LA, Dahl G, Prokop A, Ott I (2014) Eur J Med Chem 87:794
Ge R, Wang YJ, Tang L, Cheng JM, Han LG, Li YL, Li QS (2014) Appl Organomet Chem 28:204
Iqbal H, Ali S, Shahzadi S (2015) Cogent Chem 1:1029039
Dokorou V, Kovala-Demertzi D, Jasinski JP, Galani A, Demertzis MA (2004) Helv Chim Acta 87:1940
Kovala-Demertzi D, Dokorou V, Primikiri A, Vargas R, Silvestru C, Russo U, Demertzis MA (2009) J Inorg Biochem 103:738
Singh RV, Chaudhary P, Chauhan S, Swami M (2009) Spectrochim Acta Part A 72:260
Chaudhary A, Agarwal M, Singh RV (2006) Appl Organomet Chem 20:295
Sharma A, Jain A, Saxena S (2015) Appl Organomet Chem 29:499
Chauhan HPS, Shaik NM (2005) J Inorg Biochem 99:538
Bhatra P, Sharma J, Sharma RA, Singh Y (2017) Appl Organomet Chem 31:e3639
Shah FA, Sabir S, Fatima K, Ali S, Qadri I, Rizzoli C (2015) Dalton Trans 44:10467
Jain M, Gaur S, Diwedi SC, Joshi SC, Singh RV, Bansal A (2004) Phosphorus. Sulfur Silicon Relat Elem 179:1517
Sirajuddin M, Ali S, McKee V, Sohail M, Pasha H (2014) Eur J Med Chem 84:343
Nath M, Vats M, Roy P (2013) Eur J Med Chem 59:310
Watanabe M, Watanabe K (2002) Vet Hum Toxicol 44:140
Jankovics H, Pettinari C, Marchetti F, Kamu E, Nagy L, Troyanov S, Pellerito L (2003) J Inorg Biochem 97:370
Danish M, Raza MA, Ilyas T, Sharif A, Anjum N (2015) Med Chem 5:373
Awang N, Kamaludin NF, Hamid A, Mokhtar NWN, Rajab NF (2012) Pak J Biol Sci 15:833
Yang Y, Hong M, Xu L, Cui J, Chang G, Li D, Li C (2016) J Organomet Chem 804:48
Nath M, Yadav R, Gielen M, Dalil H, Vos DD, Eng G (1997) Appl Organomet Chem 11:727
Yin HD, Hong M, Li G, Wang DQ (2005) J Organomet Chem 690:3714
Prasad KS, Kumar LS, Prasad M, Revanasiddappa HD (2010) Bioinorg Chem Appl 2010:1
Tabassuma S, Khan RA, Arjmand F, Sen S, Kayal J, Juvekar AS, Zingde SM (2011) J Organomet Chem 696:1600
Kumar D, Singh VK, Khiwar SS, Saxena N (2014) J Drug Deliv Ther 4:73
Kumar D, Singh VK, Khiwar SS, Saxena N (2014) J Biomed Pharm Res 3:30
Dawara L, Singh RV (2011) App Organomet Chem 25:643
Jayabalakrishnan C, Natarajan K (2002) Trans Met Chem 27:75
Dhawan SN, Dasgupta S, Mor S, Gupta SC (1993) Ind J Heterocycl Chem 2:155
Mor S, Nagoria S (2015) Chem Biol Interface 5:389
Malhotra R, Mehta J, Puri JK (2007) Cent Eur J Chem 5:858
Srinivas P, Suresh T, Revanasiddappa M, Khasim S (2008) J Chem 5:627
Nath M, Yadav R (1997) Bull Chem Soc Jpn 70:1331
Kapoor R, Gupta A, Kapoor P, Venugopalan P (2003) Appl Organomet Chem 17:607
Buntine MA, Kosovel FJ, Tiekink ER (2003) Cryst Eng Commun 5:331
Dakternieks D, Duthie A, Smyth DR, Stapleton CP, Tiekink ER (2003) Organometallics 22:4599
Dubey RK, Baranwal P, Jha AK (2012) J Coord Chem 65:2645
Czympiel L, Lekeu JM, Hegemann C, Mathur S (2017) Inorg Chim Acta 455:197
Kashar TI (2014) Eur Chem Bull 3:878
Sallam SA, Orabi AS, El-Shetary BA, Lentz A (2002) Trans Metal Chem 27:447
Hansch C, Leo A, Unger SH, Kim KH, Nikaitani D, Lien EJ (1973) J Med Chem 16:1207
Kier LB, Hall LH (1976) Molecular connectivity in chemistry and drug research. Academic Press, New York
Kumar A, Narasimhan B, Kumar D (2007) Bioorg Med Chem 15:4113
Hyperchem 6.0 (1993) Hypercube Inc, Gainesville
TSAR 3D Version 3.3 (2000) Oxford Molecular Limited
SPSS for Windows, version 10.05 (1999) SPSS Inc, Bangalore
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Khatkar, P., Asija, S., Ahlawat, A. et al. Synthesis, characterization, in vitro antimicrobial and QSAR studies of diorganotin(IV) complexes of Schiff bases derived from 2-(3-methylbutanoyl)-1H-indene-1,3(2H)-dione and 4-substituted anilines. Monatsh Chem 150, 207–218 (2019). https://doi.org/10.1007/s00706-018-2308-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-018-2308-6