
Abstract
Synthesis of a small library of 1,4-disubstituted 1,2,3-triazoles containing benzofused N-heteroaromatic moieties was carried out through click reaction of various benzofused N-heteroaromatic alkynes with aromatic azides. All the synthesized compounds were characterized by spectroscopic techniques like IR, 1H NMR, 13C NMR, mass spectrometry and evaluated in vitro for antimicrobial activity against two Gram-positive bacteria (Bacillus subtilis, Staphylococcus aureus), one Gram-negative bacteria (Escherichia coli) and two fungi (Candida albicans, Aspergillus niger). Most of the synthesized 1,4-disubstituted 1,2,3-triazoles were found to possess significant antibacterial and antifungal activity against tested microbial species. Moreover, docking simulation of the compound 1-[(1-(4-nitrobenzyl)-1H-1,2,3-triazol-4-yl)methyl]-1H-benzo[d]imidazole was also carried out against E. coli topoisomerase II DNA gyrase B enzyme to study the binding modes and mechanism of action.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Nitrogen heterocyclic compounds play key roles in the field of medicinal chemistry [1]. Among these heterocycles, much importance has been given to the triazole nucleus which possess a wide range of pharmacological activities like antimicrobial [2, 3], antitrypanosomal [4], antimalarial [5, 6], anti-HIV [7], anticancer [8–10], antitubercular [11, 12], anticonvulsant [13], antiallergic [14], etc. The regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles through copper(I) catalyzed 1,3-dipolar cycloaddition was independently reported by Sharpless [15] and Meldal [16] in 2002, led to great success over Huisgen’s classical thermal method [17] which yields 1,4- and 1,5-disubstituted isomers. This copper(I) catalyzed 1,3-dipolar cycloaddition is one of the finest reaction of click chemistry due to better regioselectivity, versatility, and compatibility.
In addition to above clinical utility, the importance of 1,2,3-triazoles have also been recognized as proton transport agents [18], glycoside cluster arrays [19], linkers [20], dendrimers [21], hyper-branched polymers [22], and liquid crystals [23]. Likewise, benzofused N-heteroarenes such as benzimidazole [24, 25], benzotriazole [26, 27], and carbazole derivatives [28] have received considerable attention owing to their presence in many medicinal agents.
Therefore, owing to medicinal utility of above class of compounds, we have synthesized a small library of 1,4-disubstituted 1,2,3-triazoles containing benzofused N-heteroaromatic moieties. To the best of our knowledge, most of the synthesized compounds are new except 6a [29], 9a, and 9e [30]. All the synthesized compounds were characterized by spectroscopic techniques like IR, 1H NMR, 13C NMR, mass spectrometry and screened in vitro for antimicrobial activity against Bacillus subtilis, Staphylococcus aureus, Escherichia coli, Candida albicans, and Aspergillus niger. Molecular docking studies give information about interactions of the molecules with biological targets to support experimental data [31]. Therefore, to study the binding modes, docking simulation of the broadly active compound 1-[(1-(4-nitrobenzyl)-1H-1,2,3-triazol-4-yl)methyl]-1H-benzo[d]imidazole (3e) was also carried out against E. coli topoisomerase II DNA gyrase B enzyme.
Results and discussion
Chemistry
The synthetic strategy of the target compounds is given in Schemes 1, 2, and 3. Benzofused N-heterocycles, i.e., benzimidazole, benzotriazole, and carbazole were used as starting materials. The benzofused N-heteroaromatic alkynes were synthesized [32] by reacting benzimidazole, benzotriazole, or carbazole with propargyl bromide in the presence of potassium carbonate in dimethylformamide at ambient temperature. 1,4-Disubstituted 1,2,3-triazoles containing benzofused N-heteroaromatic moieties were synthesized via click reaction of benzofused N-heteroaromatic alkynes with aromatic azides which in turn were prepared by in situ reaction of aromatic bromides with sodium azide, as per literature procedure [33].



The synthesized 1,4-disubstituted 1,2,3-triazoles 3a–3f, 6a–6f, and 9a–9f were characterized by spectroscopic techniques like IR, 1H NMR, 13C NMR, and mass spectrometry. The 1H NMR spectra of the compounds 3a–3f displayed one characteristic singlet in the region at δ = 7.77–8.26 ppm due to triazolyl proton and another singlet appeared in the region at 7.84–8.33 ppm was attributed to C2-H proton of benzimidazole moiety. A singlet appeared in the range at 4.18–6.11 ppm integrating two protons which was assigned to methylene group attached to N1 of triazole ring, while methylene protons attached to C4 of the triazole ring were observed in the region at 5.40–5.59 ppm. The remaining aromatic protons were observed in the region at 6.91–8.23 ppm. In 13C NMR spectra of the compounds 3a–3f, signal due to C4 of the triazole moiety appeared in the region at δ = 142.8–143.8 ppm, whereas peak owing to C5 of triazole ring resonated in the range at 126.3–128.0 ppm. The peak observed in the region at 40.3–40.7 ppm was assigned to methylene carbon attached to N1 of benzimidazole ring, while the signal due to methylene carbon linked to N1 of the triazole ring was resonated in the region at 49.5–54.2 ppm. The carbonyl carbon of 3f appeared at 166.3 ppm. The remaining aliphatic and aromatic carbons appeared in their usual range. The IR spectra of the compounds 3a–3f exhibited absorption band in the region at 3136–3113 cm−1 due to C–H stretching vibrations of triazole ring, while C–H stretching vibrations of aromatic rings were absorbed in the range at 3089–3072 cm−1. In case of 3f, absorption band at 1720 cm−1 was attributed to carbonyl carbon. The absorption bands owing to C=C stretching vibrations of aromatic rings were observed in their common range. The mass spectra of the compounds 3a–3f showed signals because of [M+] and [M++1] ions, which are in good agreement with their calculated values.
Likewise, the 1H NMR spectra of the compounds 6a–6f displayed one characteristic singlet in the region at 7.42–8.36 ppm due to triazolyl proton. A singlet observed in the region at 5.40–5.97 ppm integrating two protons was assigned to methylene protons attached to N1 of the benzotriazole ring, while another singlet appeared in the region at 4.28–6.14 ppm was attributed to methylene protons linked to N1 of the triazole ring. Further, remaining aromatic protons were observed in the region at 6.93–8.22 ppm. In 13C NMR spectra of the compounds 6a–6f, signal due to C4 carbon atom of the triazole ring appeared in the region at 141.7–143.7 ppm, whereas C5 carbon atom absorbed in the range at 127.2–127.9 ppm. The signal appeared in the region at 49.7–54.2 ppm was assigned to methylene carbon attached to N1 of the triazole ring, while signal due to methylene carbon linked to N1 of the benzotriazole ring was observed in the region at 43.3–43.8 ppm. The carbonyl carbon of 6f appeared at 166.4 ppm. The remaining aliphatic and aromatic carbons displayed peaks in the normal range. The IR spectra of the compounds 6a–6f showed absorption band in the region at 3142–3126 cm−1 because of C–H stretching vibrations of triazole ring, while bands due to C–H stretching vibrations of aromatic rings observed in the region at 3074–3026 cm−1. In the compound 6f, band absorbed at 1720 cm−1 was assigned to carbonyl carbon of phthalimide moiety. The bands due to aromatic ring stretching vibrations were absorbed in their general range. The mass spectra of the compounds 6a–6f displayed peaks corresponding to [M+] and [M++1] ions, which are in good agreement with predicted values.
Further, 1H NMR spectra of the compounds 9a–9f exhibited characteristic peak in the region at 7.36–8.08 ppm due to triazolyl proton. A singlet corresponding to two protons in the region at 5.28–5.62 ppm was attributed to methylene protons attached to nitrogen atom of the carbazole ring, while another singlet observed in the region at 4.11–6.00 ppm integrating two protons was due to methylene group linked to N1 of triazole ring. The remaining aromatic protons appeared in the region at 6.62–8.14 ppm. In the 13C NMR spectra of the compounds 9a–9f, two characteristic signals due to C4 and C5 of the triazole moiety appeared in the region at 143.8–145.0 ppm and 126.0–126.9 ppm, respectively. The singlet appeared in the region at 38.1–39.0 ppm was attributed to methylene carbon linked to N1 of carbazole ring; whereas, the peak due to methylene carbons linked to N1 of the triazole ring resonated in the region at 49.5–54.0 ppm. The carbonyl carbon of 9f was displayed at 166.4 ppm. The remaining aliphatic and aromatic carbons resonated in their normal range. The IR spectra of the compounds 9a–9f showed absorption bands in the region at 3128–3122 and 3074–3057 cm−1 owing to C–H stretching vibrations of triazole and aromatic rings, respectively. Three strong bands were absorbed in the region at 1601–1448 cm−1 because of C=C stretching vibrations of aromatic rings. The compound 9f displayed strong absorption band at 1724 cm−1 owing to presence carbonyl carbon in the molecule. The results of mass spectral analysis of compounds 9a–9f showed presence of signals with respect to [M+] and [M++1] ions, which are in good agreement with expected values.
Antibacterial activity
The antibacterial activity of synthesized 1,4-disubstituted 1,2,3-triazoles was evaluated in vitro against two Gram-positive bacteria, i.e., B. subtilis (MTCC 441), S. aureus (MTCC 3160) and one Gram-negative bacteria, viz. E. coli (MTCC 443) by standard serial dilution method [34]. B. subtilis produces a proteolytic enzyme subtilisin, which causes ropiness. S. aureus causes boils, sties, and skin infection. E. coli, in a number of patients is responsible for gastroenteritis, urinary tract infections, and neonatal meningitis [35]. The double-strength nutrient broth was used as culture media. Dimethylsulfoxide was used as negative control. Norfloxacin, a broad spectrum antibiotic that is effective against both Gram-positive and Gram-negative bacteria was used as reference drug for antibacterial activity assay. Minimal inhibitory concentration (MIC) values of standard drug and synthesized compounds were determined in terms of µmol/cm3 × 10−2 as shown in Table 1.
Most of the synthesized compounds displayed moderate-to-good antibacterial activity against tested bacterial strains as depicted by MIC values expressed in µmol/cm3 × 10−2. Among synthesized triazoles, compounds 3e (MIC, 1.86), 3f (MIC, 1.74) and 9c (MIC, 1.70) against S. aureus while 3e (MIC, 1.86), 6e (MIC, 1.86), and 9d (MIC, 1.77) against E. coli displayed almost twofold antibacterial potency as compared to reference drug, norfloxacin. However, some of the compounds like 6b (MIC, 2.05) and 9c (MIC, 1.70) against B. subtilis; 3c (MIC, 3.94), 6c (MIC, 3.92), 6d (MIC, 4.10) and 9d (MIC, 3.54) against S. aureus; 3d (MIC, 4.12), 3f (MIC, 3.48), and 6d (MIC, 4.10) against E. coli exhibited comparable antibacterial activity to the reference drug used.
From the above results, it has been generalized that the presence of electron withdrawing group on phenyl ring improved the antibacterial activity of synthesized triazoles. Similarly, the increase in length of carbon chain at N1 position of triazole ring also enhanced the antibacterial potency of the synthesized compounds containing benzimidazolyl and carbazolyl moieties. Substitution of benzyl group by phthalimide-NCH2 at N1 position of triazole ring in the compound (3f) with benzimidazolyl moiety leads to improved bactericidal efficiency against E. coli and S. aureus.
Antifungal activity
The in vitro antifungal activity of the synthesized triazoles was tested against two fungal strains, C. albicans (MTCC 227) and A. niger (MTCC 281) by standard serial dilution method [34]. C. albicans generally causes mucosal infections in immunosuppressant patients which results in life-threatening diseases. A. niger, a xerophilic fungus also causes many serious infections in humans and other organisms [35]. Sabouraud dextrose broth was used as nutrient media while dimethylsulfoxide as a solvent control. Fluconazole, an antifungal medicine possessing triazole moiety was used as standard drug for antifungal activity assay. MIC values were reported in µmol/cm3 × 10−2 as given in Table 2.
All the synthesized compounds exhibited moderate-to-good antifungal activity against tested fungal strains as depicted by MIC values expressed in µmol/cm3 × 10−2. The compounds 6a (MIC, 2.15), 6b (MIC, 2.05), and 9e (MIC, 1.63) exhibited almost twofold antifungal activity against A. niger as compared to standard drug, fluconazole. However, in case of C. albicans, some of the compounds like 6b (MIC, 2.05) and 9b (MIC, 1.77) displayed antifungal efficacy comparable to reference drug. Further, the compounds 3b (MIC, 4.12), 3e (MIC, 3.73), 6e (MIC, 3.72), 9b (MIC, 3.54), 9c (MIC, 3.41), and 9f (MIC, 3.06) showed good antifungal activity like standard drug, against A. niger.
The results clearly indicated that the presence of electron withdrawing groups on phenyl ring improved the antifungal activity of synthesized compounds against A. niger; whereas, presence of electron donating groups on phenyl ring increased the fungicidal activity of synthesized triazoles against C. albicans. Moreover, replacement of benzyl group by phthalimide-NCH2 group at N1 position of triazole ring of compound (9f) having carbazolyl moiety increased the antifungal efficiency against A. niger.
Docking studies
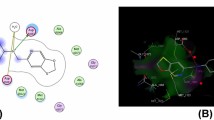
E. coli topoisomerase II DNA gyrase B enzyme was used as target for envisaging antimicrobial potential of the synthesized 1,4-disubstituted 1,2,3-triazoles containing benzofused N-heteroaromatic moiety, i.e., benzimidazole, benzotriazole, and carbazole [36, 37]. As the compounds under study contains these nuclei, docking simulations of the compound 3e (MIC, 1.86) was performed into binding site of E. coli topoisomerase II DNA gyrase B enzyme in order to find out a rational mechanism of action for antimicrobial activity of the compounds and to predict the best binding orientation of this compound. The protein of interest was taken from RCSB Protein Data Bank (PDB ID: 1KZN) and Autodock Vina docking program [38] was used for docking simulations. Best ranking binding mode of compound 3e in the active site of the enzyme showing hydrogen bonding (green dotted lines), van der Waals (green spheres), and electrostatic (magenta) interactions is depicted as two-dimensional diagram in Fig. 1. It was involved in hydrogen bond interactions with the active site residue Ser121 and Ala96. One oxygen atom of nitro group of compound 3e created a strong hydrogen bond with Ser121 (H···O distance = 2.173 Å) while another oxygen atom of nitro group a formed hydrogen bond with Ala96 (H···O distance = 2.453 Å) as depicted in Fig. 2. Several active site residues, i.e., Val43, Asp49, Ile78, Met91, Gly119, Thr165, and Val167 were involved in van der Waal’s interactions with the compound. Further, residues Asn46, Ala47, Asp73, Ile90, Val93, His95, and Val120 exhibited electrostatic interactions with the target compound. These residues except Val93, His95, Ala96, Gly119, and Ser121, also assist in fixing of co-crystallized ligand clorobiocin in the enzyme active site. For these reasons, it can be accepted that the compound under study inhibits topoisomerase II DNA gyrase B enzyme in a successful manner which could be the likely cause of their antimicrobial action. Docked conformation of compound 3e along with co-crystallized ligand clorobiocin in ribbon diagram of protein is shown in Fig. 3 and surface diagram also shown in Fig. 4.

2D schematic diagram of compound 3e docked in active site of E. coli topoisomerase II DNA Gyrase B (color figure online)

Docked pose of compound 3e showing hydrogen bonding with active site residues of E. coli topoisomerase II DNA Gyrase B

Secondary structure of E. coli topoisomerase II DNA gyrase B along with docked compound 3e

Surface diagram showing docked molecule 3e with enzyme E. coli topoisomerase II DNA Gyrase B
Conclusion
In the present case, we have synthesized a small library of 1,4-disubstituted 1,2,3-triazoles containing benzofused N-heteroaromatic moieties through copper(I)-catalyzed click reaction of benzofused N-heteroaromatic alkynes with aromatic azides. The synthesized compounds were screened in vitro for antimicrobial activity. Among synthesized compounds 3e, 3f, and 9c against S. aureus; 3e, 6e, and 9d against E. coli; 6a, 6b, and 9e against A. niger displayed almost twofold antimicrobial efficiency as compared to reference drugs. The docking simulation revealed that the compound 3e inhibits E. coli topoisomerase II DNA gyrase B enzyme through hydrogen bonding, electrostatic, and van der waal interactions. This work can be further explored for designing of new 1,4-disubstituted 1,2,3-triazoles as potential antimicrobials.
Experimental
Chemicals used in present work were purchased from Hi-media/Alfa-Aesar and were used without further purification. Solvents were dried as per standard literature procedures. Melting points of synthesized compounds were recorded in °C by applying open capillary method. The IR spectra were scanned on Shimazdu IR Affinity-I IR spectrophotometer using potassium bromide (KBr) powder and values are given in cm−1. The 1H NMR spectra were recorded on Bruker Avance II 400 MHz or Bruker 300 MHz spectrometer and 13C NMR on Bruker Avance II 400 at 100 MHz or Bruker 300 at 75 MHz, in deuterated chloroform or dimethylsulfoxide-d 6 using tetramethylsilane (TMS) as an internal standard (chemical shift in δ/ppm). Coupling constant (J) values are given in Hertz (Hz). Mass spectra were recorded on Waters Micromass Q-Tof Micro (ESI) spectrophotometer. The completion of reactions and the purity of the compounds were analyzed by thin layer chromatography (TLC) using silica gel plates (SIL G/UV254, ALUGRAM) in ethyl acetate:hexane mixture and visualized under ultraviolet lamp.
General method for the synthesis of 1,4-disubstituted 1,2,3-triazoles 3a–3f, 6a–6f, 9a–9f
The benzofused N-heteroaromatic alkynes 2, 5, and 8 were synthesized by reacting benzimidazole (1.0 mmol), benzotriazole (1.0 mmol), or carbazole (1.0 mmol) with propargyl bromide (1.2 mmol) in the presence of potassium carbonate (2.0 mmol) using dimethylformamide by continuous stirring at 10–25 °C up to 8–10 h [32]. For the synthesis of target compounds, solution of aromatic bromides (1.0 mmol) in dimethylformamide was taken in a round-bottomed flask and aqueous solution of sodium azide (3.0 mmol) was added, thereafter, stirred the reaction mixture for 1 h at 25–40 °C. To the above reaction mixture, benzofused N-heteroaromatic alkyne 2 (1.0 mmol), 5 (1.0 mmol), or 8 (1.0 mmol) was added followed by copper sulfate pentahydrate (5 mol %) and sodium ascorbate (10 mol %) [33], then, stirred the reaction contents overnight. The progress of reaction was monitored by thin layer chromatography. After the completion of reaction, ice-cold distilled water was added to the reaction mixture and product was extracted with ethyl acetate (50 cm3 × 3). The organic layer was washed with aqueous ammonia solution followed by brine solution and dried using anhydrous sodium sulfate. Filtered and evaporated the solvent under vacuum to get crude product which was further purified by column chromatography using hexane:ethyl acetate (7:3) as eluent.
1-[(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl]-1H-benzo[d]imidazole (3a, C17H15N5)
Yellowish-white solid; yield: 64 %; m.p.: 130–134 °C; 1H NMR (400 MHz, CDCl3): δ = 5.46 (s, 4H, CH2), 7.20–7.36 (m, 9H, Ar–H), 7.79 (s, 1H, C-H triazole), 7.97 (s, 1H, C2–H benzimidazole) ppm; 13C NMR (75 MHz, CDCl3): δ = 40.5, 54.2, 109.8, 120.4, 121.8, 122.4, 123.1, 127.9 (C5 triazole), 128.8, 129.1, 133.4, 134.2, 142.6, 143.2, 143.8 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3136 (C–H str., triazole ring), 3088 (C–H str., aromatic), 1656 (C=N str., aromatic), 1605, 1494, 1450 (C=C str., aromatic) cm−1; MS: m/z for C17H15N5: 290.0 [M+], 291.0 [M++1].
1-[(1-Phenethyl-1H-1,2,3-triazol-4-yl)methyl]-1H-benzo[d]imidazole (3b, C18H17N5)
Yellowish-white solid; yield: 58 %; m.p.: 122–126 °C; 1H NMR (300 MHz, CDCl3): δ = 3.11 (t, J = 10.5 Hz, 2H, CH2), 4.50 (t, J = 10.5 Hz, 2H, CH2), 5.40 (s, 2H, CH2), 6.91 (t, J = 6 Hz, 3H, Ar–H), 7.11–7.32 (m, 6H, Ar–H), 7.79 (s, 1H, C–H triazole), 7.88 (s, 1H, C2-H benzimidazole) ppm; 13C NMR (75 MHz, CDCl3): δ = 36.6, 40.5, 51.8, 109.8, 120.4, 121.8, 122.4, 123.2, 127.1 (C5 triazole), 128.5, 128.7, 133.4, 136.6, 142.4, 143.8 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3128 (C–H str., triazole ring), 3089 (C–H str., aromatic), 1656 (C=N str., aromatic), 1609, 1505, 1442 (C=C str., aromatic) cm−1; MS: m/z for C18H17N5: 304.0 [M+], 305.0 [M++1].
1-[[1-(3-Phenylpropyl)-1H-1,2,3-triazol-4-yl]methyl]-1H-benzo[d]imidazole (3c, C19H19N5)
Brown solid; yield: 72 %; m.p.: 96–98 °C; 1H NMR (300 MHz, CDCl3): δ = 2.12 (p, J = 10.5 Hz, 2H, CH2), 2.53 (t, J = 10.5 Hz, 2H, CH2), 4.18 (t, J = 10.5 Hz, 2H, CH2), 5.41 (s, 2H, CH2), 7.01–7.05 (m, 3H, Ar–H), 7.17 (t, J = 12 Hz, 3H, Ar–H), 7.24-7.28 (m, 3H, Ar–H), 7.77 (s, 1H, C–H triazole), 7.84 (s, 1H, C2–H benzimidazole) ppm; 13C NMR (75 MHz, CDCl3): δ = 31.3, 32.3, 40.7, 49.5, 109.9, 120.5, 121.8, 123.2, 126.3 (C5 triazole), 128.2, 128.5, 139.6, 142.8 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3113 (C–H str., triazole ring), 3082 (C–H str., aromatic), 1670 (C=N str., aromatic), 1615, 1525, 1438 (C=C str., aromatic) cm−1; MS: m/z for C19H19N5: 318.0 [M+], 319.0 [M++1].
1-[[1-(4-Methylbenzyl)-1H-1,2,3-triazol-4-yl]methyl]-1H-benzo[d]imidazole (3d, C18H17N5)
Light yellow solid; yield: 74 %; m.p.: 150–154 °C; 1H NMR (400 MHz, CDCl3): δ = 2.00 (s, 3H, CH3), 5.50 (s, 2H, CH2), 5.57 (s, 2H, CH2), 7.27–7.29 (m, 2H, Ar–H), 7.36 (t, J = 8.8 Hz, 3H, Ar–H), 7.40–7.43 (m, 1H, Ar–H), 7.80 (s, 1H, C–H triazole), 7.99 (s, 1H, C2–H benzimidazole), 8.17 (d, J = 8.8 Hz, 2H, Ar–H) ppm; 13C NMR (75 MHz, CDCl3): δ = 21.1, 40.5, 54.0, 109.8, 120.4, 121.6, 122.3, 123.1, 128.0 (C5 triazole), 129.8, 131.1, 138.7, 143.1, 143.7 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3118 (C–H str., triazole ring), 3072 (C–H str., aromatic), 1658 (C=N str., aromatic), 1614, 1550, 1431 (C=C str., aromatic) cm−1; MS: m/z for C18H17N5: 304.0 [M+], 305.0 [M++1].
1-[[1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl]methyl]-1H-benzo[d]imidazole (3e, C17H14N6O2)
Light yellow solid; yield: 70 %; m.p.: 144–148 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ = 5.59 (s, 2H, CH2), 5.75 (s, 2H, CH2), 7.19–7.26 (m, 2H, Ar–H), 7.50 (d, J = 8.8 Hz, 2H, Ar–H), 7.64 (t, J = 8.8 Hz, 2H, Ar–H), 8.20-8.23 (m, 2H, Ar–H), 8.26 (s, 1H, C–H triazole), 8.33 (s, 1H, C2–H benzimidazole) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ = 40.6, 52.4, 111.1, 119.9, 122.1, 122.8, 124.6, 127.1 (C5 triazole), 129.5, 131.1, 143.6, 143.8 (C4 triazole), 147.7 ppm; IR (KBr): \(\bar{v}\) = 3113 (C–H str., triazole ring), 3082 (C–H str., aromatic), 1656 (C=N str., aromatic), 1606, 1555, 1492 (C=C str., aromatic), 1523 (N–O str., asym., NO2), 1348 (N–O str., sym., NO2) cm−1; MS: m/z for C17H14N6O2: 335.0 [M+], 336.0 [M++1].
2-[[4-[(1H-Benzo[d]imidazol-1-yl)methyl]-1H-1,2,3-triazol-4-yl]methyl]isoindoline-1,3-dione (3f, C19H14N6O2)
Dark brown solid; yield: 55 %; m.p.: 152–156 °C; 1H NMR (300 MHz, CDCl3): δ = 5.45 (s, 2H, CH2), 6.11 (s, 2H, phthalimide NCH2), 7.23–7.27 (m, 3H, Ar–H), 7.73–7.77 (m, 4H, Ar–H), 7.85–7.89 (m, 2H, Ar–H), 7.99 (s, 1H, C2–H benzimidazole) ppm; 13C NMR (75 MHz, CDCl3): δ = 40.3, 49.7, 109.9, 120.4, 122.5, 123.2, 123.3, 124.2, 127.4 (C5 triazole), 131.3, 134.1, 134.8, 142.6, 143.5 (C4 triazole), 166.3 (C=O) ppm; IR (KBr): \(\bar{v}\) = 3134 (C–H str., triazole ring), 3078 (C–H str., aromatic), 1720 (C=O str.), 1654 (C=N str., aromatic), 1612, 1492 (C=C str., aromatic) cm−1; MS: m/z for C19H14N6O2: 359.0 [M+], 360.0 [M++1].
1-[(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl]-1H-benzo[d][1,2,3]triazole (6a, C16H14N6)
Light brown crystalline solid; yield: 74 %; m.p.: 156–160 °C (Ref. [29] 158–160 °C).
1-[(1-Phenethyl-1H-1,2,3-triazol-4-yl)methyl]-1H-benzo[d][1,2,3]triazole (6b, C17H16N6)
Light yellow solid; yield: 70 %; m.p.: 120–124 °C; 1H NMR (400 MHz, CDCl3): δ = 3.12 (t, J = 7.2 Hz, 2H, CH2), 4.51 (t, J = 7.2 Hz, 2H, CH2), 5.91 (s, 2H, CH2), 6.93 (d, J = 8.4 Hz, 2H, Ar–H), 7.07–7.16 (m, 4H, Ar–H), 7.37 (t, J = 8.4 Hz, 1H, Ar–H), 7.47 (s, 1H, C–H triazole), 7.66 (d, J = 8.4 Hz, 1H, Ar–H), 8.04 (d, J = 8.4 Hz, 1H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 36.6, 43.8, 51.9, 110.1, 119.9, 123.2, 124.1, 127.2 (C5 triazole), 127.6, 128.5, 132.6, 136.6, 141.7 (C4 triazole), 146.2 ppm; IR (KBr): \(\bar{v}\) = 3142 (C–H str., triazole ring), 3026 (C–H str., aromatic), 1612, 1494, 1450 (C=C str., aromatic) cm−1; MS: m/z for C17H16N6: 305.0 [M+], 306.0 [M++1].
1-[[1-(3-Phenylpropyl)-1H-1,2,3-triazol-4-yl]methyl]-1H-benzo[d][1,2,3]triazole (6c, C18H18N6)
Creamy white solid; yield: 72 %; m.p.: 92–96 °C; 1H NMR (400 MHz, CDCl3): δ = 2.19 (p, J = 7.2 Hz, 2H, CH2), 2.60 (t, J = 7.2 Hz, 2H, CH2), 4.28 (t, J = 7.2 Hz, 2H, CH2), 5.97 (s, 2H, CH2), 7.08 (d, J = 7.2 Hz, 2H, Ar–H), 7.16 (d, J = 7.2 Hz, 1H, Ar–H), 7.25 (t, J = 7.2 Hz, 2H, Ar–H), 7.36 (t, J = 7.2 Hz, 1H, Ar–H), 7.45 (s, 1H, C–H triazole), 7.48 (d, J = 7.2 Hz, 1H, Ar–H), 7.71 (d, J = 8.4 Hz, 1H, Ar–H), 8.04 (d, J = 8.4 Hz, 1H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 31.4, 32.4, 43.8, 49.7, 110.1, 119.9, 122.8, 124.2, 126.4, 127.7 (C5 triazole), 128.4, 128.6, 132.7, 139.8, 142.1 (C4 triazole), 146.2 ppm; IR (KBr): \(\bar{v}\) = 3134 (C–H str., triazole ring), 3070 (C–H str., aromatic), 1604, 1494, 1448 (C=C str., aromatic) cm−1; MS: m/z for C18H18N6: 319.0 [M+], 320.0 [M++1].
1-[[1-(4-Methylbenzyl)-1H-1,2,3-triazol-4-yl]methyl]-1H-benzo[d][1,2,3]triazole (6d, C17H16N6)
Light brown solid; yield: 70 %; m.p.: 152–156 °C; 1H NMR (400 MHz, CDCl3): δ = 2.32 (s, 3H, CH3), 5.40 (s, 2H, CH2), 5.92 (s, 2H, CH2), 7.09–7.15 (m, 4H, Ar–H), 7.35 (t, J = 8.0 Hz, 1H, Ar–H), 7.42 (s, 1H, C–H triazole), 7.46 (t, J = 8.0 Hz, 1H, Ar–H), 7.71 (d, J = 8.0 Hz, 1H, Ar–H), 8.01 (d, J = 8.0 Hz, 1H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 21.2, 43.8, 54.2, 110.2, 119.8, 122.7, 124.1, 127.0, 127.6 (C5 triazole), 128.5, 131.0, 132.7, 139.0, 142.3 (C4 triazole), 146.1 ppm; IR (KBr): \(\bar{v}\) = 3134 (C–H str., triazole ring), 3072 (C–H str., aromatic), 1614, 1508, 1438 (C=C str., aromatic) cm−1; MS: m/z for C17H16N6: 305.0 [M+], 306.0 [M++1].
1-[[1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl]methyl]-1H-benzo[d][1,2,3]triazole (6e, C16H13N7O2)
Light yellow solid; yield: 76 %; m.p.: 220–226 °C; 1H NMR (400 MHz, DMSO-d 6 ): δ = 5.76 (s, 2H, CH2), 6.07 (s, 2H, CH2), 7.41 (t, J = 7.2 Hz, 1H, Ar–H), 7.50–7.58 (m, 3H, Ar–H), 7.91 (d, J = 8.4 Hz, 1H, Ar–H), 8.05 (d, J = 8.4 Hz, 1H, Ar–H), 8.22 (d, J = 8.4 Hz, 2H, Ar–H), 8.36 (s, 1H, C–H triazole) ppm; 13C NMR (100 MHz, DMSO-d 6 ): δ = 43.3, 52.4, 111.3, 119.6, 124.4, 125.1, 127.9 (C5 triazole), 129.5, 133.1, 142.5, 143.7 (C4 triazole), 145.7, 147.7 ppm; IR (KBr): \(\bar{v}\) = 3126 (C–H str., triazole ring), 3074 (C–H str., aromatic), 1602, 1556, 1445 (C=C str., aromatic), 1523 (N–O str., asym., NO2), 1344 (N–O str., sym., NO2) cm−1; MS: m/z for C16H13N7O2: 336.0 [M+], 337.0 [M++1].
2-[[4-[(1H-benzo[d][1,2,3]triazol-1-yl)methyl]-1H-1,2,3-triazol-1-yl]methyl]isoindoline-1,3-dione (6f, C18H13N7O2)
Light yellow solid; yield: 55 %; m.p.: 148–152 °C; 1H NMR (400 MHz, CDCl3): δ = 5.95 (s, 2H, CH2), 6.14 (s, 2H, phthalimide NCH2), 7.34 (t, J = 8.4 Hz, 1H, Ar–H), 7.45 (t, J = 8.4 Hz, 1H, Ar–H), 7.75–7.89 (m, 5H, Ar–H), 7.92 (s, 1H, C–H triazole), 8.00 (d, J = 7.6 Hz, 1H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 43.5, 49.8, 110.2, 119.8, 123.6, 124.0, 124.2, 127.7 (C5 triazole), 131.3, 132.7, 135.0, 142.7 (C4 triazole), 146.1, 166.4 (C=O) ppm; IR (KBr): \(\bar{v}\) = 3126 (C–H str., triazole ring), 3026 (C-H str., aromatic), 1720 (C=O str.), 1608, 1456 (C=C str., aromatic) cm−1; MS: m/z for C18H13N7O2: 360.0 [M+], 361.0 [M++1].
9-[(1-Benzyl-1H-1,2,3-triazol-4-yl)methyl]-9H-carbazole (9a, C22H18N4)
White solid; yield: 78 %; m.p.: 158–162 °C (Ref. [30] 160–162 °C).
9-[(1-Phenethyl-1H-1,2,3-triazol-4-yl)methyl]-9H-carbazole (9b, C23H20N4)
Dull-white solid; yield: 62 %; m.p.: 132–136 °C; 1H NMR (300 MHz, CDCl3): δ = 2.96 (t, J = 7.0 Hz, 2H, CH2), 4.30 (t, J = 7.0 Hz, 2H, CH2), 5.51 (s, 2H, CH2), 6.62 (t, J = 8.0 Hz, 1H, Ar–H), 6.80 (t, J = 8.0 Hz, 2H, Ar–H), 7.01–7.04 (m, 3H, Ar–H), 7.20 (t, J = 8.0 Hz, 2H, Ar–H), 7.36 (s, 1H, C–H triazole), 7.36–7.39 (m, 3H, Ar–H), 8.05 (d, J = 8.0 Hz, 2H, Ar–H) ppm; 13C NMR (75 MHz, CDCl3): δ = 36.6, 38.7, 51.6, 108.7, 119.4, 120.4, 121.7, 123.1, 125.9, 126.9 (C5 triazole), 128.6, 136.6, 140.0, 143.8 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3124 (C–H str., triazole ring), 3064 (C–H str., aromatic), 1593, 1490, 1448 (C=C str., aromatic) cm−1; MS: m/z for C23H20N4: 353.0 [M+], 354.0 [M++1].
9-[[1-(3-Phenylpropyl)-1H-1,2,3-triazol-4-yl]methyl]-9H-carbazole (9c, C24H22N4)
White solid; yield: 66 %; m.p.: 140–146 °C; 1H NMR (400 MHz, CDCl3): δ = 2.07 (p, J = 7.2 Hz, 2H, CH2), 2.49 (t, J = 7.2 Hz, 2H, CH2), 4.11 (t, J = 7.2 Hz, 2H, CH2), 5.62 (s, 2H, CH2), 6.90–7.45 (m, 11H, Ar–H), 7.46 (s, 1H, C–H triazole), 8.10 (d, J = 7.6 Hz, 2H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 31.3, 32.3, 39.0, 49.5, 108.8, 119.5, 120.3, 120.5, 121.4, 123.2, 126.0 (C5 triazole), 128.4, 128.6, 139.9, 140.1, 144.5 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3122 (C–H str., triazole ring), 3062 (C–H str., aromatic), 1597, 1489, 1452 (C=C str., aromatic) cm−1; MS: m/z for C24H22N4: 367.0 [M+], 368.0 [M++1].
9-[[1-(4-Methylbenzyl)-1H-1,2,3-triazol-4-yl]methyl]-9H-carbazole (9d, C23H20N4)
White solid; yield: 58 %; m.p.: 158–162 °C; 1H NMR (400 MHz, CDCl3): δ = 2.28 (s, 3H, CH3), 5.28 (s, 2H, CH2), 5.57 (s, 2H, CH2), 7.01 (d, J = 7.6 Hz, 3H, Ar–H), 7.07 (d, J = 7.6 Hz, 2H, Ar–H), 7.20–7.26 (m, 2H, Ar–H), 7.41–7.43 (m, 4H, Ar–H), 8.07 (d, J = 8.0 Hz, 2H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 21.1, 39.0, 54.0, 108.8, 119.4, 120.4, 121.3, 123.1, 126.0 (C5 triazole), 128.0, 129.7, 131.4, 138.6, 140.1, 144.8 (C4 triazole) ppm; IR (KBr): \(\bar{v}\) = 3122 (C–H str., triazole ring), 3074 (C–H str., aromatic), 1597, 1485, 1450 (C=C str., aromatic) cm−1; MS: m/z for C23H20N4: 353.0 [M+], 354.0 [M++1].
9-[[1-(4-Nitrobenzyl)-1H-1,2,3-triazol-4-yl]methyl]-9H-carbazole (9e, C22H17N5O2)
Dull-yellow solid; yield: 60 %; m.p.: 148–152 °C (Ref. [30] 150–153 °C).
2-[[4-[(9H-Carbazol-9-yl)methyl]-1H-1,2,3-triazol-1-yl]methyl]isoindoline-1,3-dione (9f, C24H17N5O2)
Brown solid; yield: 56 %; m.p.: 154–158 °C; 1H NMR (400 MHz, CDCl3): δ = 5.57 (s, 2H, CH2), 6.00 (s, 2H, phthalimide NCH2), 7.12–7.25 (m, 3H, Ar–H), 7.40–7.48 (m, 3H, Ar–H), 7.70–7.75 (m, 2H, Ar–H), 7.80–7.82 (m, 2H, Ar–H), 7.99 (d, J = 7.6 Hz, 1H, Ar–H), 8.05 (d, J = 7.6 Hz, 2H, Ar–H) ppm; 13C NMR (100 MHz, CDCl3): δ = 38.7, 49.6, 108.8, 119.9, 120.3, 122.5, 123.1, 126.2 (C5 triazole), 131.3, 134.8, 139.3, 145.0 (C4 triazole), 166.4 (C=O) ppm; IR (KBr): \(\bar{v}\) = 3128 (C–H str., triazole ring), 3057 (C–H str., aromatic), 1724 (C=O), 1601, 1456 (C=C str., aromatic) cm−1; MS: m/z for C24H17N5O2: 408.0 [M+], 409.0 [M++1].
Antibacterial activity
All the synthesized triazoles were screened for their in vitro antibacterial activity against two Gram-positive bacteria, i.e., B. subtilis (MTCC 441), S. aureus (MTCC 3160) and one Gram-negative bacteria, E. coli (MTCC 443) by standard serial dilution method [34] using a stock solution of 100 µg/cm3. Dimethylsulfoxide was employed as a solvent control. Dilutions of test compounds were prepared in double-strength nutrient broth. One cm3 nutrient broth was taken in each of seven test tubes. To the first test-tube 1.0 cm3 of drug solution (100 µg/cm3) was added aseptically to get the concentration of 50 µg/cm3. From this dilution, other concentrations were prepared by serial dilution to get final concentrations of 25, 12.5, 6.25, 3.12, 1.56, 0.78 µg/cm3 in test-tube number two to seven. All the test tubes were aseptically inoculated by 0.1 cm3 of desired bacterial strain in sterile saline. The inoculated test samples were then incubated at 37 ± 1 °C for 1 day. Norfloxacin, a broad spectrum antibiotic, was used as standard drug and also tested under similar experimental conditions for comparison with synthesized triazoles.
Antifungal activity
The in vitro antifungal activity of synthesized triazoles was performed against two fungal strains, i.e., C. albicans (MTCC 227) and A. niger (MTCC 281) by serial dilution method [34] using a stock solution of 100 µg/cm3 concentration of compounds. Sabouraud dextrose broth was used as a fungal culture media and DMSO as solvent control. One cm3 of freshly prepared sterile culture media was added aseptically in each test tube followed by serial dilution with synthesized compounds to prepare concentrations of 25–0.78 µg/cm3. Further these dilutions of triazole compounds were inoculated with 0.1 cm3 of suspension of respective microorganism contained in sterilized saline. Then samples of compounds loaded with microorganisms incubated at 27 ± 1 °C for 2 days in case of C. albicans and at 25 ± 1 °C for 7 days in case of A. niger. Fluconazole, a widely accepted antifungal drug, was used as standard drug.
Computational details
The docking procedure chosen in the present study was in accordance with the procedure given by Kumar [39–41]. Structures of the compounds were sketched with Mavin Sketch 5.10 [42] which were optimized and cleaned with gradient optimization. The X-ray crystallographic structure of E. coli topoisomerase II DNA gyrase B enzyme along with co-crystallized ligand CBN (PDB ID: 1KZN) was obtained from Brookhaven Protein Databank (http://www.rcsb.org/pdb). Preparation of protein was accomplished with UCSF Chimera 1.9 [43]. Incomplete side chains were completed with Dunbrack rotamer library [44] and Gasteiger charges were assigned with Antechamber [45]. Structures of ligands and proteins were transformed into pdbqt format with the aid of AutoDock tools [46]. Docking simulations were carried out by AutoDock Vina program. The Vina search space taken was center_x = 19.7572768649, center_y = 30.6958566405, center_z = 36.3020605554, size_x = 25.0, size_y = 24.2096062606, size_z = 21.3564769495. The exhaustiveness for docking was set to be 8. Validation of docking protocols was made by means of reported crystal structure of protein–ligand complex. Protocols selected for the AutoDock Vina docking studies were realistic to mimic the X-ray structure as the root-mean square deviation (RMSD) between the conformations of the CBN from the X-ray crystal structure and that from AutoDock Vina was less than 2 Å. These protocols were used for docking of the compound under study into the active site of DNA Gyrase B. Results were pictured with the help of PyMol [47] and Discovery Studio version 3.5 [48].
References
Dua R, Shrivastava S, Sonwane SK, Srivastava SK (2011) Advan Biol Res 5:120
Kaushik CP, Kumar K, Singh SK, Singh D, Saini S (2013) Arab J Chem. doi:10.1016/j.arabjc.2013.09.023
Gaur M, Goel M, Sridhar L, Ashok TDS, Prabhakar S, Dureja P, Raghunathan P, Eswaran SV (2012) Monatsh Chem 143:283
Junqueira GG, Carvalho MR, Andrade P, Lopes CD, Carneiro ZA, Sesti-Costa R, Silva JS, Carvalho I (2014) J Braz Chem Soc 25:1872
Asif M (2014) Int J Adv Res Chem Sci 1:22
Pereira GR, Brandao GC, Arantes LM, Oliveira HA, Paula RC, Nascimento MFA, Santos FM, Rocha RK, Lopes JCD, Oliveira AB (2014) Eur J Med Chem 73:295
Silva F, Souza MCBV, Frugulhetti IIP, Castro HC, Souza SL, Souza TM, Rodrigues DQ, Souza AMT, Abreu PA, Passamani F, Rodrigues CR, Ferreira VF (2009) Eur J Med Chem 44:373
Singh P, Raj R, Kumar V, Mahajan MP, Bedi PM, Kaur T, Saxena AK (2012) Eur J Med Chem 47:594
Elamari H, Slimi R, Chabot GG, Quentin L, Scherman D, Girard C (2013) Eur J Med Chem 60:360
Lal K, Kaushik CP, Kumar K, Kumar A, Qazi AK, Hamid A, Jaglan S (2014) Med Chem Res 23:4761
Jordao AK, Sathler PC, Ferrreia VF, Campos VR, Souza MCBV, Castro HC, Lannes A, Lourenco A, Rodrigues CR, Bello ML, Lourenco MCS, Carvalho GSL, Almeida MCB, Cunha AC (2011) Bioorg Med Chem 19:5605
Yempala T, Sridevi JP, Yogeeswari P, Sriram D, Kantevari S (2014) Eur J Med Chem 71:160
Karakurt A, Aytemir MD, Stables JP, Ozalp M, Betul FK, Ozbey S, Dalkara S (2006) Arch Pharm 339:513
Buckle DR, Rockell CJM, Smith H, Spicer BA (1986) J Med Chem 29:2262
Rostovtsev VV, Green LG, Fokin VV, Sharpless KB (2002) Angew Chem Int Ed 41:2596
Tornøe CW, Charistensen C, Meldal M (2002) J Org Chem 67:3057
Huisgen R (1963) Angew Chem 75:604
Nagamani C, Versek C, Thorn M, Tuominen MT, Thayumanavan TS (2010) J Polym Sci 48:1851
Dondoni A, Marra A (2006) J Org Chem 71:7546
Madhuri V, Kumar VA (2012) Nucleosides, Nucleotides Nucleic Acids 31:97
Haridas V, Lal K, Sharma YK (2007) Tetrahedron Lett 48:4719
Li ZA, Yu G, Hu P, Ye C, Liu YQ, Qin JG, Li Z (2009) Macromolecules 42:1589
Balamurugan S, Yeap GY, Mahmood WAK (2014) Liq Cryst 41:776
Khokra SL, Choudhary D (2011) Asian J Biochem Pharm Res 1:476
Singh G, Kaur M, Chander M (2013) Int Res J Pharm 4:82
Jamkhandi CM, Disouza JI (2012) Asian J Biochem Pharm Res 2:123
Ren Y, Zhang L, Zhou CH, Geng RX (2014) Med Chem 4:640
Fousteris MA, Papakyriakou A, Koutsourea A, Manioudaki M, Lampropoulou E, Papadimitriou E, Spyroulias GA, Nikolaropoulos SS (2008) J Med Chem 51:1048
Verkhozina ON, Kizhnyaev VN, Vereshchagin LI, Rokhin AV, Smirnov AI (2003) Russ J Org Chem 39:1792
Surineni G, Yogeeswari P, Sriram D, Kantevari S (2015) Med Chem Res 24:1298
Harmsen RAG, Sivertsen A, Michetti D, Brandsdal BO, Sydnes LK, Haug BE (2013) Monatsh Chem 144:479
Barbosa FCG, Oliveira RN (2011) J Braz Chem Soc 22:592
Farooq T, Haung BK, Sydnes LK, Tornroos KW (2012) Monatsh Chem 143:505
Cappucino JG, Sherman N (1999) Microbiology: a laboratory manual, 4th edn. Addison Wesley, Longman Inc, Harlow, p 263
Mor S, Mohil R, Kumar D, Ahuja M (2012) Med Chem Res 21:3541
Chinnadurai S, Mahalingam R, Perumal R, Vaithiyanathan S, Devadasan V (2014) Can Chem Trans 2:248
Kaushik CP, Lal K, Kumar A, Kumar S (2014) Med Chem Res 23:2995
Trott O, Olson AJ (2010) J Comput Chem 31:455
Kumar A, Kumar S, Jain S, Kumar P, Goyal R (2013) Med Chem Res 22:5431
Lal K, Kumar A, Pavan MS, Kaushik CP (2012) Bioorg Med Chem Lett 22:4353
Lal K, Kaushik CP, Kumar A (2015) Med Chem Res. doi:10.1007/s00044-015-1378-9
Mavin Sketch 5.10.1 (2012) ChemAxon Ltd. http://www.chemaxon.com
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) J Comput Chem 25:1605
Dunbrack RL (2002) Curr Opin Struct Biol 12:431
Wang J, Wang W, Kollman PA, Case DA (2006) J Mol Graph Model 25:247
AutoDock Tools, version 1.5.6 rc2 (2010) Stefano Forte. Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute. http://mgltools.scripps.edu
PyMol(TM) Molecular Graphics System Version 0.99rc6 (2006) Delano Scientific LLC. http://www.pymol.org/funding.html
Discovery Studio v4.0 client (2014) Accelrys Software Inc
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kaushik, C.P., Kumar, K., Lal, K. et al. Synthesis and antimicrobial evaluation of 1,4-disubstituted 1,2,3-triazoles containing benzofused N-heteroaromatic moieties. Monatsh Chem 147, 817–828 (2016). https://doi.org/10.1007/s00706-015-1544-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-015-1544-2