
Abstract
In this research, the pyridoacridines alkaloids have been docked computationally to the active site of Topoisomerase II using four PDB structures (1PVG, 1QZR, 1AJ6 and 1ZXM). iGEMDOCK 2.1, AutoDock Vina 1.1.2 and AutoDock 4.2.1 were employed to perform the automated molecular docking. The results of docking studies generated docking scores and IC50 values. Moreover, 3D pictures of ligand enzyme complexes afforded valuable data regarding the binding orientation of each inhibitor in the active site of Topoisomerase II.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Topoisomerases are the nuclear enzymes that induce transient breaks in the DNA. There are two types of Topoisomerases—Topoisomerase I and Topoisomerase II. Topoisomerase I seems not to be as essential as Topoisomerase II for the survival of eukaryotic cells (Caroline and Katherine, 1998; Smiley et al., 2007; Thakur, 2011). So, it has been reviewed that most attention has been paid on the drugs acting on Topoisomerase II (Molinski, 1993; Hangstler et al., 2002). This enzyme plays a critical role in transcription and replication of DNA (Cortés et al., 2003; Kumar and Rawat, 2011), and also maintains the DNA topology, distangles knotted DNA, maintains correct chromosome condensation, decondensation, and segregation (Sorensen et al., 1996; Wang, 1998). It has been well-reviewed that it is a validated target of various antineoplastic drugs like anthracyclines (doxorubicin, daunorubicin), epipodophyllotoxins but are limited by their tumor resistance mechanism, side effects profile and also by their sensitivity to P-gp receptor mediated efflux. Now it is well-established that several antineoplastic agents those act through intercalation also acts on Topoisomerase II (Lee, 1996, Hawtin et al., 2010).
Literature of traditionally occurring medicines shows that natural products have very wide role and they are valuable source for new drug discovery (Fabricant and Farnsworth, 2001; Butler, 2004; Harvey, 2008). Crystallographic data based molecular modeling has been used to aid the design of synthetic analogs of natural products (Corbett and Berger, 2004; Huang et al., 2011; Nematollahi et al., 2011). There are so many compounds possessing pyridoacridine skeleton having anti-HIV activity, Ca2+ releasing activity, metal chelating property, DNA intercalating activity, and Topoisomerase II inhibition property (Bhakuni and Rawat, 2005; Kumar and Rawat, 2011). Pyridoacridines are colored marine alkaloids having 7H- pyrido [2, 3, 4-kl] acridine skeleton (Molinski, 1993; Skyler and Heathcock, 2002) (Fig. 1).

General structure of pyridoacridines
The tetracyclic members of this class are archetypical pyridoacridines. Nine cytotoxic tetracyclic alkaloids, Cystodytins A-I have been identified from yellow tunicate Cystodytes dellechiajei. Cystodytins A and Cystodytins F are shown in Fig. 2a, b Cystodytins A–C are the first member of this class (Bontemps et al., 2010; Kumar and Rawat 2011). Cystodytin A–C and Varamine A and C (Fig. 2c, d) have been found to be cytotoxic against L-1210 (Kumar and Rawat, 2011).

a–h Examples of various novel pyridoacridine alkaloids
A novel pentacyclic alkaloid, ascididemin isolated from brown colored tunicate Didemnum sp. has been found to be cytotoxic against L-1210 marine leukemia cells (Kumar and Rawat, 2011). Ascididemin and its isomers also exhibited cytotoxicity against U-87MG, U-373MG, T-47D, MCF-7, HCT-15, A-549, A-427, T-24, and J-82 cell lines (Matsumoto et al., 2003; Bhakuni and Rawat, 2005) (Fig. 2e, f).
Cyclodercitin, a hexacyclic alkaloid shown in Fig. 2g, obtained from the extracts of a deep violet sponge Dercitus sp. inhibits the proliferation of P-388 murine leukemia cells. It has also been reviewed that Eilatin, a heptacyclic pyridoacridine exhibits cytotoxic activity against HCT cell line (Stanslas et al., 2000; Kumar and Rawat, 2011). Eilatin octacyclic analog does not show any activity against HT-29 (Fig. 2h).
Therefore, it can be said that mostly all pyridoacridines have an immense role as anticancer agents. It is supposed that these compounds show anticancer effect due to inhibition of Topoisomerase II (Dias et al., 2005; Sanchez-Carrasco et al., 2008; Cragg et al., 2009). So, we report herein the study describing binding of pyridoacridine alkaloids against Topoisomerase II, which has been carried out by molecular docking investigations.
Computational details
Regarding this issue the crystal structures of Topoisomerase II were obtained from the Brookhaven Protein Data Bank http://www.rcsb.org/pdb (PDB entry: 1ZXM, 1PVG, 1AJ6 and 1QZR). To carry out docking studies, the 2D structures of various pyridoacridine ligands (Kumar and Rawat, 2011; Menna et al., 2011) were drawn (Fig. 3a–c) and these were converted into 3D and their energy was minimized using MM2 method with RMS gradient of 0.1 centers. These compounds were saved in mdl mol and pdb files for further use. Docking studies were carried out by iGEMDOCK 2.1, AutoDock Vina 1.1.2, and AutoDock 4.2.1 programs. In order to perform the task, the various interactions formed by docked ligands were observed.



a–c Structures of various pyridoacridine ligands
To insure that the ligand orientation and the position obtained from the docking studies were likely to represent valid and reasonable binding modes of the inhibitors, docking of co-crystallized ligands were carried out for all protein structures (1PVG, 1QZR, 1AJ6, and 1ZXM). The ligand conformation found in the crystal structure, was extracted and docked back to the corresponding binding pocket. Results of control docking showed the optimal orientation of the docked inhibitor, close to that of the original orientation found in the crystal as shown in Fig. 4. The RMS deviation of less than 0.2 Å between the docked and crystal ligand coordinates indicate very good alignment of the experimental and calculated positions.

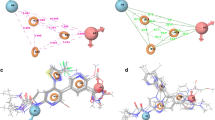
ANP docked molecule (1PVG) (UCSF Chimera ver. 1.5.3)
In iGEMDOCK (iGEMDOCK ver. 2.1), drug screening was used as default settings with population size 200, 70 generations, and 3 numbers of solutions. iGEMDOCK scoring function was chosen along with ligand intra energy with hydrophobic and electrostatic preference both as 1.
Finally, ranking of compounds were done by pharmacological energy i.e.,
whereas, E GEMDOCK is the docked energy of iGEMDOCK and E(E)pharma, E(H)pharma, and E(V)pharma are the pharmacological scores of electrostatics, hydrogen-bonding, and vdW interactions (Hsu et al., 2011), respectively.
For AutoDock Vina (Trott and Olson, 2010), ligands were removed from pdb files and protein molecules were prepared by deleting solvent molecules and non-complex ions. Incomplete side chains were replaced using Dun Brack Rotamer library (Dunbrack, 2002). Hydrogens were added and gasteiger charges were calculated using Antechamber. The prepared files were saved in pdb format and used for further studies. Similarly, ligand files were prepared in pdb format with explicit hydrogen addition. All pdb files were transformed into pdbqt format. Grid center was placed on the active site. The sizes and centers of grid box are given in Table 1.
Exhaustiveness which influences the thoroughness of the global search algorithm was set to be 8. Then, finally docking results were viewed using PDB and PDBQT files.
To gain better insight, AutoDock (Morris et al., 1999) was also employed to dock the selected pyridoacridine ligands. The prepared ligand files were transformed to pdbqt format, non-polar hydrogens were merged and charges were defined. The grid calculations were setup and maps were calculated using the program AutoGrid. The Grid maps were centered on the ligand binding site and dimensions were noted. The grid spacing was 0.3750 Å and the default AutoDock parameter settings were used for docking. The grid centers and number of points are shown in Table 2. All docking runs were performed using Lamarckian genetic algorithm and the obtained Dock scores were reported in kcal/mol. The docking protocol utilized in the study consisted of 10 independent GA runs, using an initial population of 150 randomly placed individuals, a maximum number of 250,000 energy evaluation, mutation rate of 0.02, a crossover rate of 0.80, and an elitism value of 1.
Results and discussion
Topoisomerase II inhibitors with varying structural features and inhibition constants were selected from the literature and were docked into the catalytic site of Topoisomerase II. Dock runs of pyridoacridine ligands on protein 1ZXM, 1PVG, 1QZR, and 1AJ6 using iGEMDOCK, AutoDock, and Auto Dock Vina resulted in few best compounds that were evaluated based on their binding compatibility [docked energy (kcal/mol)] with the receptor. The results of docking experiments with these inhibitors are summarized in Tables 3 and 4. These results are mainly evaluated by structure analysis of the docked complexes.
The IC50 values (μM) were recorded for the lowest binding energy mode by AutoDock Tools (AutoDock Tools ver. 1.5.6 rc2). The calculated IC50 values could not be correlated with the experimental values as the later values are not from direct inhibition of Topoisomerase II. The predicted IC50 value and experimental IC50 values are shown in Table 5.
Hydrogen-bonding interactions of compounds were visualized using Discovery Studio Visualizer as shown in Fig. 5. Compound no. 6 and 7 are having hydroxyl group which showed hydrogen-bonding interactions with SER 149, ASN 150, ALA 167, and LYS 168. Compound No. 56 was sandwiched in between ARG 76 and ILE 78 through Sigma-Pi stacking interactions (Fig. 6a). Docked poses of some compounds showing hydrogen-bonding interaction and Van der Waal interactions are shown with the help of AutoDock Tools in Fig. 7. Most of the compounds showed hydrogen-bonding interactions with SER 128, SER-149, TYR 165, ALA 167, ILE 120, ARG 76, and Vander Waal interactions with ASN-91, ALA-92, ASN-95, ARG-98, ILE-125, ILE-141, PHE-142, and THR-215. Surface diagram of all the ligands docked on PDB structure 1QZR is shown in Fig. 6b (PyMol, 2008).

a, b H-bonding interactions of compound no. 16 and 12 with Topoisomerase II (1QZR and 1PVG)

a The Sigma–Pi interactions of compound no. 56 with Topoisomerase II (1AJ6) and b Surface diagram of all docked molecules into Topoisomerase II (1QZR)

a, b) Docking poses, interaction of compound no. 3 and 5 with Topoisomerase II (1AJ6 and 1ZXM)
The compound no. 11 exhibits a good score with iGEMDOCK but not with other two programs and its predicted IC50 value is found to be very high as compared to experimental value which may be due to some physical properties of the molecule and/or some another mechanism responsible for anticancer activity. For molecules containing lesser bulkier groups attached to the ring, good scores were given by all the three programs.
The correlation of docking scores by the two programs AutoDock and AutoDock Vina is shown graphically in Fig. 8. It can be seen that most of the compounds shows a correlation in their docking scores e.g., Compound no. 14 and 15 but there are also some compounds like 46 and 47, which do not correlate in their docking scores. Almost all the compounds except 11, 22, and 35 gave better score then bounded ligand with AutoDock Vina and AutoDock in case of PDB 1AJ6. Scores of iGEMDOCK could not be correlated with docking scores of other two softwares used.

Graphical representation of docking scores by the two docking programs Autodock and Autodock Vina
Conclusion
Docking programs allowed us to estimate the docking scores, binding modes, and inhibition constants for the molecules under study. Almost all the compounds chosen except a few are found to be active against Topoisomerase II. In case of PDB 1AJ6, some ligands showed better fitness even than the co-crystallized ligand. An idealized representation of each ligand that makes every possible potential interaction with the binding site and other data obtained from all three programs iGEMDOCK, AutoDock Vina, and AutoDock, conclude that pyridoacridines are successfully docked into the protein binding site. Furthermore, this study will help in designing of novel derivatives of pyridoacridines and in discovery of new chemical entities for anticancer therapy.
References
iGEMDOCK (version 2.1) BioXGEM Lab © 2001–2010
AutoDock Tools (version 1.5.6 rc2), Stefano Forte. Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute http://mgltools.scripps.edu © (1999–2010)
Bhakuni DS and Rawat DS (2005) Bioactive marine natural products. Springer, New York, ISBN: 1-4020-3472-5
Bontemps N, Bry D, López-Legentil S (2010) Structures and antimicrobial activities of pyridoacridine alkaloids isolated from different chromotypes of the ascidian Cystodytes dellechiajei. J Nat Prod 73:1044–1048
Butler MS (2004) The role of natural product chemistry in drug discovery. J Nat Prod 67:2141–2153. doi:10.1021/np040106y
Caroline AA, Katherine LM (1998) Eukaryotic DNA topoisomerase II β. BioEssays 20:215–226
Corbett KD, Berger JM (2004) Structure, molecular mechanisms, and evolutionary relationships in DNA Topoisomerases. Annu Rev Biophys Biomol Struct 33:95–118. doi:10.1146/annurev.biophys.33.110502.140357
Cortés F, Pastor N, Mateos S, Dominguez I (2003) Roles of DNA topoisomerases in chromosome segregation and mitosis. Mutat Res 543:59–66
Cragg GM, Grothaus PG, Newman DJ (2009) Impact of natural products on developing new anti-cancer agents. Chem Rev 109:3012–3043
Dias N, Vezin H, Lansiaux A et al (2005) Topoisomerase inhibitors of marine origin and their potential use as anticancer agents. Top Curr Chem 253:89–108. doi:10.1007/b100444
Discovery Studio Visualizer (version 2.5.5.9350), Accelrys Software Inc. © 2005–2009
Dunbrack RL Jr (2002) Rotamer libraries in the 21st century. Curr Opin Struct Biol 12:431–440
Fabricant DS, Farnsworth NR (2001) The value of plants used in traditional medicine for drug discovery. Environ Health Perspect 109:69–75
Hangstler JG, Heimerdinger CK, Schiffer IB et al (2002) Dietary Topoisomerase II- poisons contribution of soy products to infant leukaemia. EXCLI J 1:8–14
Harvey AL (2008) Natural products in drug discovery. Drug Discov Today 13:894–3426. doi:10.1016/j.drudis.2008.07.004
Hawtin RE, Stockett DE, Byl JAW et al (2010) Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II. PLOS one 5:1–3
Hsu KC, Chen YF, Lin SR, Yang JM (2011) iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis. BMC Bioinformatics 12:1–11
Huang XY, Shan ZJ, Zhai HL et al (2011) Molecular design of anticancer drug leads based on three-dimensional quantitative structure_activity. J Chem Inf Model 51:1999–2006
Kumar D, Rawat DS (2011) Marine natural alkaloids as anticancer agents. In: Opportunity challenge and scope of natural products in medicinal chemistry, pp 213–268. ISBN: 978-81-308-0448-4
Lee KH (1996) Anticancer drug design based on plant-derived natural products. J Biomed Sci 6:236–250
Matsumoto SS, Biggs J, Copp BR (2003) Mechanism of ascididemin-induced cytotoxicity. Chem Res Toxicol 16:113–122
Menna M, Fattorusso E, Imperatore C (2011) Alkaloids from marine ascidians. Molecules 16:8694–8732. doi:10.3390/molecules16108694
Molinski TF (1993) Marine pyridoacridine alkaloids: structure, synthesis and biological chemistry. Chem Rev 93:1825–1838
Morris GM, Goodshell DS, Huey R, Hart WE, Halliday RS, Belew RK, Olson AJ, Autodock (1999) (version 4.2.1), Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institutes, http://www.scripps.edu/pub/olson-web/doc/Autodock/), La Jolla, CA, USA
Nematollahi A, Aminimoghadamfarouj N, Wiart C (2011) Design and Modeling Studies on Liriodenine derivatives as novel topoisomerase II Inhibitors. Int J Chem Tech Res 3:1622–1627
PyMol (TM) (2008) Evaluation Product Delano Scientific LLC. http://www.pymol.org/funding.html © 2008
Sanchez-Carrasco S, Delcros JG, Moya-Garcia AA et al (2008) Study by optical Spectroscopy and molecular dynamics of the interaction of acridine-spermine conjugate with DNA. Biophys Chem 133:54–65. doi:10.1016/j.bpc.2007.12.003
Skyler D, Heathcock CH (2002) The pyridoacridine family tree: a useful scheme for designing synthesis and predicting undiscovered natural products. J Nat Prod 65:1573–1581
Smiley RD, Collins TRL, Hammes GG, Hsieh TS (2007) Single-molecule measurements of the opening and closing of the DNA gate by eukaryotic topoisomerase II. PNAS 104:4840–4845
Sorensen BS, Sinding J, Andersen AH, Alsner J, Jensen PB, Westergaard O (1996) Mode of action of topoisomerase II targeting agents at a specific DNA sequence. Uncoupling the DNA binding, cleavage and relegation events. J Mol Biol 228:778–786
Stanslas J, Hagan DJ, Ellis MJ et al (2000) Antitumor polycyclic acridines. 7.1 synthesis and biological properties of DNA affinic tetra- and pentacyclic acridines. J Med Chem 43:1563–1572
Thakur DS (2011) Topoisomerase II Inhibitors in cancer treatment. IJPSN 3:1173–1181
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
UCSF Chimera (version 1.5.3) by the Regents of the University of California © 2000–2011
Wang JC (1998) Moving one DNA double helix through another by a type II DNA topoisomerase: the story of a simple molecular machine. Q Rev Biophys 31(2):107–144
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kumar, A., Kumar, S., Jain, S. et al. Study of binding of pyridoacridine alkaloids on topoisomerase II using in silico tools. Med Chem Res 22, 5431–5441 (2013). https://doi.org/10.1007/s00044-013-0496-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-013-0496-5