
Abstract
A highly anti-diastereoselective three-component Mannich reaction of aromatic amines and aromatic aldehydes with cyclohexanone in the presence of silica-supported ferric hydrogensulfate has been developed. The best selectivity was obtained where there were electron-donating groups on both aldehyde and amine. Selectivity decreases when electron-withdrawing groups are present on the aldehyde; in these cases selectivity is improved if an electron-donating group is present on the amine.
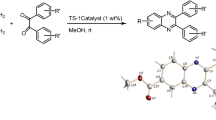
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Mannich reaction is one of the most important methods for construction of carbon–carbon bonds to build β-aminocarbonyl compounds [1–5]. These compounds are useful precursors for synthesis of β-lactams [3, 6, 7], α [8–11] and γ-aminoalcohols [13], α and β-amino acid derivatives [13], peroxy acetylenic alcohols/ethers [4], and medicinally important materials [1].
Several strategies are available for diastereoselective synthesis of β-aminocarbonyl compounds, including organocatalysis [6, 7, 12–21], transition-metal catalysis [8–11, 22–25], Bronsted and Lewis acid catalysis [26–38], phase-transfer catalysis [39–41], HPA catalysis [42, 43], biocatalysis [44], and ionic-liquid catalysis [45, 46]. Organocatalytic asymmetric Mannich reactions are the most important approach to the direct anti-enantioselective reaction of aldehydes and ketones [47, 48].
Iron is an important metal in living systems and is a sustainable metal catalyst for performing a wide range of different chemical transformations. Iron salts have often been used in organic synthesis, for example oxidation, reduction, coupling reactions, and cycloaddition, because they are inexpensive, nontoxic, readily available, easily recyclable, and environmentally benign [49, 50].
Therefore, to achieve diastereoselective synthesis of β-aminoketones via a three-component Mannich reaction, we chose ferric hydrogensulfate (FHS) as catalyst. Recently we have successfully used FHS for nucleophilic addition of nucleophiles to aldehydes [51–53]. In this work, we performed nucleophilic addition of enols to aldimines. Herein we report, for the first time, the iron-salt-catalyzed three-component Mannich reaction of aromatic aldehydes and aromatic amines with cyclohexanone to afford Mannich products with high anti-diastereoselectivity.
Results and discussion
We selected the three-component Mannich reaction of aniline (1 eq), benzaldehyde (1 eq), and cyclohexanone (1.2 eq) as model reaction to optimize the reaction conditions. When we used FHS (10 mol %) as catalyst in ethanol, moderate diastereoselectivity (anti:syn = 67:33) was obtained.
Furthermore reduction of catalyst molar ratio did not change the diastereoselectivity of the reaction significantly (Table 1, entries 2 and 3). Other catalysts, for example FeCl2 and FeCl3·6H2O (Table 1, entries 10 and 14) also gave moderate diastereoselectivity. With Mn(HSO4)2 as catalyst the percentage of the anti isomer improved to 75 % (Table 1, entry 9). When we used FHS supported on silica (1:9) as catalyst better diastereoselectivity was observed. The best result (>99 % anti) was obtained when the molar ratio of catalyst to starting material was approximately 10 mol % (Table 1, entry 6).

As the results in of Table 1 show, silica alone improves the selectivity of the reaction to 84 % anti isomer compared with the catalyst free and solid state reactions (Table 1, entries 4, 5, 12). However when FHS is supported on silica using the same conditions the reaction time drops from 2 h to 30 min and diastereoselectivity increases from 84 % to more than 99 %. According to Table 1, when the reaction is carried out under catalyst-free conditions in ethanol, diastereoselectivity decreases to 60:40 ratio (Table 1, entry 4).
We believe diastereoselectivity depends on different factors, for example solvent, silica, and iron salt. When the ratio of ferric hydrogensulfate to silica gel was 9:1, catalytic activity was highest. However, when NaHSO4 was used instead of Fe(HSO4)3, with the same molar ratio, diastereoselectivity decreased to 72:28. This observation shows that besides the solvent and silica gel, the iron cation has a significant effect on the diastereoselectivity of the reaction.
When the reaction conditions had been optimized for the model reaction, we screened aromatic aldehydes and aromatic amines in reactions with cyclohexanone. As the results in Table 2 show, with benzaldehyde itself the only stereoisomer obtained is anti, except with 4-Cl aniline which gives 80 % anti isomer (Table 2, entries 1–5). Weak electron-withdrawing or electron donating groups on benzaldehyde, for example 4-chloro and 4-methyl, respectively, do not change the diastereoselectivity of the reaction. However, the presence of a strong electron-withdrawing group (EWG), for example NO2, at the para or meta positions of the benzaldehyde destroys the diastereoselectivity of the reaction. Interestingly, with these EWGs on the aldehyde, some diastereoselectivity is observed when an electron-donating group (EDG) is present on the aromatic amine (Table 2, compounds 16 and 19). As expected, the presence of substitution in the ortho position of aniline reduces the nucleophilicity of the compound and thus reaction times increase substantially (Table 2, compounds 2, 6, and 11).

The efficiency of our catalyst was then tested with other ketones as substrates (Table 3). In an initial series of experiments, a representative set of ketones, including both cyclic and acyclic substrates, were reacted with benzaldehyde and aniline in the presence of 10 mol % catalyst. Acyclic ketones required longer reaction times for complete conversion, but regioselectivity was very high. Cyclopentanone behaved similar to cyclohexanone and gave exclusively the anti product.

The most plausible mechanism is imine formation between amine and aldehyde followed by nucleophilic addition to the imine of the enol formed by the catalyst. The most probable transition states, which explain the diastereoselectivity of the reaction, are shown in Fig. 1.

Probable transition state structures
We believe the catalyst is mostly involved in the second step of the mechanism. Although it can assist enol formation and nucleophilic addition of enol to aldimine, it mostly controls the diastereoselectivity of the reaction by controlling the stereochemistry of the transition state. As shown in Fig. 1, transition states A and B are more favorable sterically. These transition states will give the anti isomer. Transition states C and D are highly hindered and so unlikely to be formed in the presence of catalyst. This highly energetic transition state will lead to the syn isomer. According to the proposed mechanism the anti product is expected from the cis imine (Fig. 1, B), and, because of the steric effect, the anti product is more likely to be formed from the trans imine (Fig. 1, A and C).
Electronic effects of substituents on the aldehyde and amine effect the stability of these transition states. Whereas EWGs on the aldehyde reduce the stability of the transition state and thus destroy the diastereoselectivity of the reaction, EDGs on the amine compensate for the EWGs and increase the diastereoselectivity of the reaction.
We propose that the transition state is chair like and both oxygen and nitrogen coordinate to the iron center (Fig. 2). When the nitro group is located on benzaldehyde the nitrogen of the imine will not coordinate well with the iron center, and thus the steric effect will not determine the diastereoselectivity of the products.

Electronic effect of the transition state
The modified transition state illustrated in Fig. 2 shows that the electronic effect is a major factor affecting the diastereoselectivity of the product. When a methyl group is present at the para position of the aniline diastereoselectivity increases. In fact, with imines containing 4-methyl substitution on the aniline ring, the electron density of transition state depicted in Fig. 2 improves and chelation of nitrogen to the iron center will occur more efficiently and diastereoselectivity proceeds toward anti (Table 2, compounds 16 and 19).
In conclusion, we present in this paper a completely diastereoselective three-component Mannich reaction catalyzed by ferric hydrogensulfate supported on silica gel. The advantages of our method are the high yield and excellent selectivity of the reaction, and the inexpensive and heterogeneous catalyst.
Experimental
All solvents and reagents were purchased from Merck and Fluka. The silica for preparation of the supported catalyst was 230 mesh for column chromatography and was purchased from Merck. NMR spectra were recorded on Bruker Aspect 3000 (100 MHz) and Bruker Avance (400 MHz) spectrometers. All chemical shifts are reported as ppm and were referenced to residual solvent signals. IR spectra were recorded on a ThermoNicolet Avatar-370-FTIR spectrometer.
Preparation of silica ferric hydrogensulfate (10 mol %)
Ferric hydrogensulfate (5 mmol) and silica gel 230 mesh for column chromatography (45 mmol) were placed in a mortar and the mixture was ground for 5 min. The mixture was then placed in a 50 cm3 flask, 25 cm3 absolute ethanol was added, and the mixture was stirred at room temperature for 10 h. The mixture was then filtered and the residue was dried at 100 °C for 2 h. A white homogeneous powder was obtained which was stored in a desiccator.
Typical procedure for synthesis of β-aminocyclohexanones
To a mixture of aromatic amine (1 mmol), aromatic aldehyde (1 mmol), and cyclohexanone (1.2 mmol) in 1 cm3 ethanol, 88.7 mg silica ferric hydrogensulfate (10 mol %) was added. The reaction mixture was stirred at room temperature and the progress of the reaction monitored by TLC. After completion of the reaction, 2 cm3 methanol was added, followed by dropwise addition of water until the product began to precipitate. The mixture was then filtered by suction and the residue was washed with 0.5 cm3 methanol and 0.5 cm3 petroleum ether. The crude product was extracted from the precipitate by washing with CHCl3. The solution was dried over Na2SO4 then the solvent was removed. The solid product obtained was suitable for spectroscopic application. Further purification was performed by crystallization from aqueous ethanol.
2-[(4-Chlorophenyl)[(2-methylphenyl)amino]methyl]cyclohexanone (6, C20H22ClNO)
Yield 54 %; m.p.: 106–107 °C; 1H NMR (400 MHz, CDCl3): δ = 1.70-1.90 (m, 3H), 1.90-2.10 (m, 3H), 2.27 (s, 3H), 2.30-2.50 (m, 2H), 2.80-2.90 (m, 1H), 4.67 (d, 1H, J = 6 Hz, anti), 6.37 (d, 1H, J = 8 Hz), 6.65 (t, 1H, J = 7.5 Hz), 6.98 (t, 1H, J = 7.2 Hz), 7.08 (d, 1H, J = 5.6 Hz), 7.34 (AB-q, 4H) ppm; 13C NMR (100 MHz, CDCl3): δ = 17.68, 23.90, 27.92, 31.63, 42.06, 57.50, 57.57, 110.65, 117.29, 126.90, 128.64, 128.69, 128.74, 130.14, 131.60, 140.51, 144.92, 212.95 ppm; IR (KBr): \( \bar{\nu } \) = 3,374 (s, NH), 3,029 (w), 2,945 (s), 1,702 (s, C=O), 1,605 (m), 1,518 (s), 1,449 (m), 1,314 (m), 826 (m), 744 (m) cm−1.
2-[(4-Methylphenyl)[(2-methylphenyl)amino]methyl]cyclohexanone (11, C21H25NO)
Yield 60 %, m.p.: 114–115 °C; 1H NMR (400 MHz, CDCl3): δ = 1.65-1.85 (m, 2H), 1.86-2.00 (m, 4H), 2.23 (s, 3H), 2.33 (s, 3H), 2.30-2.50 (m, 2H), 2.75-2.85 (m, 1H), 4.65 (d, 1H, J = 7.2 Hz, anti), 4.60-4.80 (br, NH), 6.41 (d, 1H, J = 7.6 Hz), 6.60 (t, 1H, J = 7.2 Hz), 6.95 (t, 1H, J = 7.2 Hz), 7.05 (d, 1H, J = 7.2 Hz), 7.13 (d, 2H, J = 7.6 Hz), 7.28 (d, 2H, J = 8 Hz) ppm; 13C NMR (100 MHz, CDCl3): δ = 17.67, 21.13, 23.42, 27.92, 31.21, 41.68, 57.69, 57.73, 110.65, 116.93, 122.55, 126.85, 127.08, 129.24, 129.99, 136.78, 138.76, 145.16, 213.53 ppm; IR (KBr): \( \bar{\nu } \) = 3,402 (m, NH), 3,382 (m, NH), 3,014 (w), 2,945 (m), 1,701 (s, C=O), 1,604 (m), 1,518 (s), 1,450 (m), 823 (m), 745 (m) cm−1.
2-[(4-Methylphenyl)[(3-methylphenyl)amino]methyl]cyclohexanone (12, C21H25NO)
Yield 72 %; m.p.: 124–125 °C; 1H NMR (400 MHz, CDCl3): δ = 1.60-1.82 (m, 2H), 1.82-2.10 (m, 4H), 2.25 (s, 3H), 2.35 (s, 3H), 2.35-2.55 (m, 2H), 2.70-2.80 (m, 1H), 4.65 (d, 1H, J = 7.2 Hz, anti), 6.39 (d, 1H, J = 7.6 Hz), 6.44 (s, 1H), 6.50 (d, 1H, J = 7.6 Hz), 7.00 (t, 1H, J = 7.6 Hz), 7.16 (d, 2H, J = 7.6 Hz), 7.31 (d, 2H, J = 8 Hz) ppm; 13C NMR (100 MHz, CDCl3): δ = 21.13, 21.48, 23.53, 27.96, 31.20, 41.70, 57.58, 57.62, 110.50, 114.57, 118.44, 127.15, 128.98, 129.21, 130.55, 136.70, 138.77, 147.33, 213.12 ppm; IR (KBr): \( \bar{\nu } \) = 3,359 (s, NH), 3,051 (w), 2,942 (m), 1,702 (s, C = O), 1,605 (s), 1,533 (m), 1,305 (m), 821 (m), 782 (m) cm−1.
2-[[(4-Methylphenyl)amino](3-nitrophenyl)methyl]cyclohexanone (19, C20H22N2O3)
Yield 85 %; m.p.: 136–137 °C; 1H NMR (400 MHz, CDCl3): δ = 1.50-1.90 (m, 3H), 1.90-2.20 (m, 3H), 2.21 (s, 3H), 2.30-2.50 (m, 2H), 2.85-2.95 (m, 1H), 4.73 (d, 0.7H, J = 5.2 Hz, anti), 4.86 (d, 0.3H, J = 4.4 Hz, syn), 6.48 (d, 2H, J = 8.4 Hz), 6.93 (d, 2H, J = 8 Hz), 7.49 (t, 1H, J = 8 Hz), 7.80 (d, 1H, J = 7.2 Hz), 8.09 (d, 1H, J = 8 Hz), 8.28 (s, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ = 20.38, 20.40, 24.46, 24.99, 27.21, 27.85, 29.23, 31.98, 42.41, 42.53, 56.37, 57.12, 57.33, 57.91, 113.69, 114.21, 122.23, 122.36, 122.54, 127.31, 127.55, 129.32, 129.38, 129.72, 129.80, 133.72, 134.25, 144.36, 144.66, 148.41, 210.90 (syn), 212.03 (anti) ppm; IR (KBr): \( \bar{\nu } \) = 3,383 (s, NH), 2,932 (m), 1,704 (C=O, syn), 1.697 (C=O, anti), 1,616 (m), 1,518 (s), 1,346 (s), 811 (s), 711 (m) cm−1.
References
Arend M, Westermann B, Risch N (1998) Angew Chem Int Ed 37:1044
Notz W, Tanaka F, Barbas CF III (2004) Acc Chem Res 37:580
Cordova A (2004) Acc Chem Res 37:102
Dikusar EA, Kozlov NG, Popova LA, Moiseichuk KL, Yuvchenko AP (2003) Russ J Gen Chem 73:625
Kobayashi S, Mori Y, Fossey JS, Salter MM (2011) Chem Rev 111:2626
Lou S, Taoka BM, Ting A, Schaus SE (2005) J Am Chem Soc 127:11256
Hayashi Y, Tsuboi W, Ashimine I, Urushima T, Shoji M, Sakai K (2003) Angew Chem Int Ed 42:3677
Ishitani H, Ueno M, Kobayashi S (1997) J Am Chem Soc 119:7153
Kobayashi S, Hamada T, Manabe K (2002) J Am Chem Soc 124:5640
Matsunaga S, Kumagai N, Harada S, Shibasaki M (2003) J Am Chem Soc 125:4712
Trost BM, Terrell L (2003) J Am Chem Soc 125:338
Ibrahem I, Zou W, Engqvist M, Xu Y, Cόrdova A (2005) Chem Eur J 11:7024
Zhang H, Mifsud M, Tanaka F, Barbas CF III (2006) J Am Chem Soc 128:9630
Zheng X, Quan Y, Wang Y (2010) Eur J Org Chem 515
Zhang H, Chuan Li YZ, Peng Y (2009) Adv Synth Catal 351:2288
Ramasartry SSV, Zhang H, Tanaka F, Barbas CF III (2007) J Am Chem Soc 129:288
List B (2000) J Am Chem Soc 122:9336
List B, Pojarliev P, Biller WT, Martin H (2002) J Am Chem Soc 124:827
Notz W, Tanaka F, Watanabe S, Chowdari NS, Turner JM, Thyumanavan R, Barbas CF III (2003) J Org Chem 68:9624
Chowdari NS, Suri JT, Barbas CF III (2004) Org Lett 6:2507
Cheong PH, Zhang H, Thayumanavan R, Tanaka F, Houk KN, Barbas CF III (2006) Org Lett 8:811
Yanagisawa A, Saito H, Harada M, Arai T (2005) Adv Synth Catal 347:1517
Qiu R, Yin S, Zhang X, Xia J, Xu X, Luo S (2009) Chem Commun 4759
Kureshy RI, Agrawal S, Saravan S, Khan NH, Shah AK, Abdi SHR, Bajaj HC, Suresh E (2010) Tetrahedron Lett 51:489
Xia J, Qiu R, Yin S, Zhang X, Luo S, Au C, Xia K, Wong W (2010) J Organomet Chem 695:1487
Uraguchi D, Tereda M (2004) J Am Chem Soc 126:5356
Blatt AH, Gross N (1964) J Org Chem 29:3306
Manabe K, Kobayashi S (1999) Org Lett 1:1965
Iimura S, Nobutou D, Manabe K, Kobayashi S (2003) Chem Commun 1644
Wu Y, Cai J, Hu Z, Lin G (2004) Tetrahedron Lett 45:8949
Akiyama T, Matsuda K, Fuchibe K (2005) Synlett 322
Guo Q, Liu H, Quo C, Luo S, Gu Y, Gong L (2007) J Am Chem Soc 129:3790
Lu G, Cai C (2010) Catal Commun 11:745
Ranu BC, Samanta S, Guchheit SK (2002) Tetrahedron 58:983
Loh T, Chen S (2002) Org Lett 4:3647
Ollevier T, Nadeau E, Guay-Bégin A (2006) Tetrahedron Lett 47:8351
Eftekhari-Sis B, Abdollahifar A, Hashemi MM, Zirak M (2006) Eur J Org Chem 5152
Hong D, Yang Y, Wang Y, Liu X (2009) Synlett 1107
Ooi T, Kameda M, Fujii T, Maruoka K (2004) Org Lett 6:2397
Shen W, Wang L, Tiam H (2008) J Fluorine Chem 129:267
Jafari AA, Moradgholi F, Tamaddon F (2009) Eur J Org Chem 1249
Azizi N, Torkiyan L, Saidi M (2006) Org Lett 8:2079
Das B, Kumar AS, Kanth BR (2009) Synth Commun 39:3111
He T, Li K, Wu M, Fang X, Wang N, Wang H, Li C, Lu X (2010) J Mol Catal B Enzyme 67:189
Gong K, Fang D, Wang H, Liu Z (2007) Monatsh Chem 138:1195
Fang D, Gong K, Zhang D, Liu Z (2009) Monatsh Chem 140:1325
Alza E, Rodriguez-Escrich C, Sayalero S, Bastero A, Pericas MA (2009) Chem Eur J 15:10167
Martin-Rapun R, Fan X, Sayalero S, Bahramnejad M, Cuevas F, Pericas MA (2011) Chem Eur J 17:8780
Plietker B (2008) Iron catalysis in organic chemistry. Wiley-VCH, Weinheim
Enthaler S, Junge K, Beller M (2008) Angew Chem Int Ed 47:3317
Eshghi H, Bakavoli M, Moradi H (2008) Chin Chem Lett 19:1423
Rahimizadeh M, Eshghi H, Bakhtiarpoor Z, Pordel M (2009) J Chem Res 269
Eshghi H, Alipour A, Damavandi S (2011) Synth React Inorg Metal-Org Nano-Metal Chem 41:266
Kozlov NS, Vorobeva GV (1968) Vestsi Akad Navuk BSSR. Ser Khim Navuk 4:107
Kidwai M, Mishra NK, Bansal V, Kumar A, Musomdar S (2009) Tetrahedron Lett 50:1355
Sharghi H, Jokar M (2009) Can J Chem 88:14
Kumar V, Sharma U, Verma PK, Kumar N, Singh B (2011) Chem Pharm Bull 59:639
Nemati F, Fakhaei FS, Amoozadeh A, Hayeniaz YS (2011) Synth Commun 41:3695
Yi W-B, Cai C (2006) J Fluorine Chem 127:1515
Bigdeli M, Nemati F, Mahdavinia G (2007) Tetrahedron Lett 48:6801
Wu H, Chen X-M, Wan Y, Ye L, Xin H-Q, Xu H–H, Yue C-H, Pang L–L, Ma R, Shi D-Q (2009) Tetrahedron Lett 50:1062
Xia J, Qiu R, Yin S, Zhang X, Luo S, Au C-T, Xia K, Wong W-Y (2010) J Organomet Chem 695:1487
An Y-J, Wang C–C, Liu Z-P, Tao J-C (2012) Helv Chim Acta 95:43
Acknowledgments
We are grateful to Ferdowsi University of the Mashhad Research Council for their financial support of this work (grant: 3/16044).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
1H/13C NMR and FTIR spectra of all compounds are available as supporting information free of charge via the internet.
Rights and permissions
About this article
Cite this article
Eshghi, H., Rahimizadeh, M., Hosseini, M. et al. Diastereoselective three-component Mannich reaction catalyzed by silica-supported ferric hydrogensulfate. Monatsh Chem 144, 197–203 (2013). https://doi.org/10.1007/s00706-012-0800-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-012-0800-y