
Abstract
Secondary α-deuterium kinetic isotope effects confirm that [2+4] cycloaddition between (E)-2-phenylnitroethene and cyclopentadiene occurs in concerted manner, on both the pathway leading to 6-endo-phenyl-5-exo-nitronorbornene and the pathway leading to the corresponding 6-exo-phenyl-5-endo-nitro isomer. According to Hammond terminology the transition states on competitive pathways should be considered in terms of symmetrical early states.
Graphical abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The [2+4] cycloadditions of simple nitroalkenes with conjugated dienes proceed according to a concerted mechanism, as confirmed by kinetic studies and by stereospecificity observed in the reactions [1–4]. However, the results are insufficient to assess symmetry and the degree of formation of new σ bonds in the postulated transition complexes. Therefore, pursuing our studies on reactivity of nitroalkenes in [2+4] π-electron cycloadditions [5–9], we decided to explore the α-secondary kinetic isotope effect (SKIE). For our investigations we selected (E)-2-phenylnitroethenes (1a–1d) with varied degree of deuteration at the nitrovinyl moiety as the 2π-electron components, and cyclopentadiene (2) as the 4π-electron component. Recently [10], we have confirmed by means of high-performance liquid chromatography (HPLC) and 1H nuclear magnetic resonance (NMR) spectroscopy that in nitromethane the [2+4] cycloaddition of these reactants is stereoselective and leads to 6-endo-phenyl-5-exo-nitro- (3a–3d) and 6-exo-phenyl-5-endo-nitronorbornenes (4a–4d) in quantitative yields (Scheme 1).

Based on the combination of femtosecond time-resolved observation of the intermediates [11] and quantum-chemical simulation of the reaction pathways [12], analysis of SKIEs [13, 14] gives excellent insight into the symmetry of the transition states, knowledge which is vital for better understanding of the underlying reaction mechanism. It has been known [1, 2, 4, 15] for some time now that, in the case of extremely π-deficient dienophiles, a two-stage mechanism with a zwitterionic intermediate or a one-step two-stage mechanism, involving formation of a heterodiene, may compete with the concerted mechanism. This was confirmed by experimental studies [16–20] and quantum-chemical calculations [21–25].
Results and discussion
The α-secondary deuterium kinetic isotope effects result from changes in oscillation behavior of C–H bonds not participating directly in the chemical reaction, upon substitution of one of the hydrogen atoms with its heavier isotope [13, 14]. In the cycloaddition in question, these are the C–H bonds in the nitrovinyl moiety of phenylnitroethene 1a. During the course of the reaction, the hybridization of Cα and Cβ atoms of the moiety changes from trigonal to tetrahedral. This leads to increase of the frequency of out-of-plane bending of Cα–H and Cβ–H bonds, which is closely related to the system zero-point energy (ZPE). The difference between the ZPE of substrates and the ZPE of transition complexes is lower for D-substituted dienophiles than for H-substituted dienophile [14]. Therefore, in the case of a concerted mechanism, the rate constant (k D) of the [2+4] cycloaddition of phenylnitroethenes 1b–1d with cyclopentadiene should be higher than the corresponding rate constant (k H) of the reaction of 1a. However, in the case of a two-stage process, one of the k D values should be equal to k H. When the k H/k D ratio (SKIE) is known, the degree of rehybridization of Cα and Cβ atoms in the transition state may be estimated, and consequently information can be obtained about the degree of formation of new σ bonds and the transition state symmetry.
The data in Table 2 show that the SKIE, which results from substitution of the hydrogen atom at Cα position of dienophile 1a with deuterium, is low. In particular, the SKIE is 0.95 for the reaction 1b + 2 → 3b (pathway A) and 0.94 for the reaction 1b + 2 → 4b (pathway B). If it is assumed that the configuration of the Cα atom is fully tetrahedral within the transition state, the frequency of out-of-plane bending of the Cα–H bond should increase from 970 cm−1 [26] to about 1,430 cm−1 [10], which corresponds to SKIE of about 0.7 [14, 27]. Therefore, the SKIE data indicate that, in both transition states, the configuration at the Cα atom is much closer to the trigonal configuration found in nitroalkene 1a than to the tetrahedral configuration found in the cycloadducts 3a and 4a. The Cα atom of the dienophile substructure tends to adopt the tetrahedral configuration of the C-6 atom in the cycloadduct. However, the conjugation of π bonds of the phenyl ring and the nitrovinyl moiety tends to retain the configuration of the dienophile. Both tendencies compete within the two structures.
When the hydrogen atom at Cβ position of dienophile 1a is substituted with deuterium, SKIE is 0.95 for both reactions 1c + 2 → 3c and 1c + 2 → 4c. This confirms that the rehybridization of the reaction site is not far advanced. The competition of two opposite tendencies is also visible in this case. The Cβ atom of the dienophile substructure tends to adopt the tetrahedral configuration of the C-5 atom in the cycloadduct. However, the conjugation of the nitro group with the vinylidene fragment of the nitroalkene restrains that process.
As could be expected on the basis of the rule of geometric mean [28], the SKIE values for the reactions 1d + 2 → 3d and 1d + 2 → 4d are very close to the products of the corresponding SKIE values obtained for the reactions 1b + 2 and 1c + 2, confirming the reliability of our results.
The presented data indicate that rehybridization of the Cα and Cβ atoms of the nitroethenyl moiety of the dienophile is not very advanced in both stereoisomeric transition states. Hence, the transition states of the reaction studied can be interpreted as synchronous early transition states.
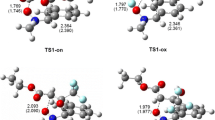
The results obtained are consistent with quantum-chemical calculations. In particular, our B3LYP/6-31G* simulation of the reaction paths has confirmed that: (1) the reaction proceeds by a concerted mechanism, (2) the distances between the reaction sites in both transition states differ only insignificantly (Fig. 1), and (3) the calculated SKIE values correlate well with the experimental data (Table 2). Moreover, it should be noted that B3LYP/6-311G** calculations also confirm the one-step mechanism of the reaction under study. Detailed analysis of the quantum-chemical results is the subject of a separate paper [29].

Transition structures approximated with B3LYP/6-31G* method for [2+4] cycloaddition of (E)-2-phenylnitroethene to cyclopentadiene in nitromethane (for simulation of the solvent effect the PCM approach was applied)
Conclusions
The [2+4] cycloaddition of (E)-2-phenylnitroethene and cyclopentadiene occurs in concerted manner, on both the path leading to 6-endo-phenyl-5-exo-nitronorbornene and the path leading to the corresponding 6-exo-phenyl-5-endo-nitro stereoisomer. The π deficiency of the dienophile [30] is not sufficient to induce a two-step mechanism. On the basis of degree of rehybridization of the reaction sites, the transition states involved in both competing reaction paths should be considered as symmetrical early states.
Experimental
Reagents
(E)-2-phenylnitroethenes (1a–1d) and cyclopentadiene (2) were synthesized according to known procedures [31–33]. Their purity was confirmed by means of HPLC and 1H NMR spectroscopy. Pure-grade nitromethane (POCh, Gliwice, Poland) was used as a solvent; it was dried over 4-Ǻ molecular sieves and distilled before use.
Kinetic procedure
The rates of the cycloaddition reactions were followed by HPLC using the integrated area (A) of the peak corresponding to the cycloadducts 3 and 4. A Knauer system (Smartline 1000 HPLC pump and Smartline 4000 thermostat) equipped with Smartline 2500 UV–Vis detector and LiChrospher 100-10 RP column (4 × 240 mm i.d.) was applied for analysis. Methanol–water (50:50 v/v) at flow rate of 1.3 cm3/min was used as the eluent. Analyses were carried out at 25 °C and λ = 254 nm. At these conditions both cycloadducts had the same retention times (t = 11.3 min). The starting reaction mixtures were prepared by adding a weighed quantity of suitable (E)-2-phenylnitroethene to the solution of freshly distilled cyclopentadiene in dry nitromethane. The initial concentration of the latter reactant was 0.05 mol/dm3, whereas the former one was always used in 20–22-fold molar excess. The mixtures were placed in ampoules of 1 cm3 capacity and thermostated at 70 ± 0.2 °C. During kinetic runs, the ampoules were taken periodically from the thermostat and immediately cooled in an ice bath. Samples of 250 mm3 were taken from the ampoules with a microsyringe and were diluted with 1,000 mm3 methanol. The solutions were analyzed by HPLC at analytical wavelength 210 nm. It was found that, for this wavelength, the Bouguer–Beer plot was linear within the concentration range studied. Second-order rate constants k total (Table 1) were obtained by a routine method [34]. Control experiments showed that, under the conditions used for the kinetic measurements, the concentrations of 3 and 4 were measured with an error less than 3%. After completion of the reaction, the ratio of the cycloadducts in product mixture, γ = [3]/[4], was determined by HPLC. The analyses were carried out at 5 °C, using LiChrospher 100-5 RP column (4 × 240 mm i.d.), methanol–water (55:45 v/v) mixture as eluent, and eluent flow rate of 0.5 cm3/min. At these conditions, the peaks corresponding to the cycloadducts were clearly separated [t(3) = 203 min, t(4) = 224 min]. The k total and γ values were converted to the rate constants k A and k B (Table 2) according to the following formulas:
References
Perekalin VV, Lipina ES, Berestovitskaya VM, Efremov DA (1994) Nitroalkenes; conjugated nitro compounds. Wiley, Chichester
Ono N (2001) In: The nitro group in organic synthesis. Wiley, New York
Baer HH, Urbas L (1970) In: The chemistry of nitro and nitroso groups, Part 2, Wiley, New York
Jasiński R, Kwiatkowska M, Barański A (2007) Wiad Chem 67:485
Barański A (2002) J Phys Org Chem 15:78
Jasiński R, Wąsik K, Mikulska M, Barański A (2009) J Phys Org Chem 22:717
Jasiński R, Barański A (2008) Collect Czech Chem Commun 73:649
Jasiński R, Jasińska E, Barański A (2008) Chem Heterocyclic Comp 44:735
Jasiński R, Barański A (2006) Pol J Chem 80:1493
Kwiatkowska M (2008) PhD thesis, Cracow University of Technology, Cracow
Zewail HA (1996) J Phys Chem A 100:12701
Cramer CJ (2002) Essentials of computational chemistry. Wiley, Chichester
Meander L, Saunders WH (1980) Reaction rates of isotopic molecules. Wiley, New York
Mattsson O, Westway KC (1998) Adv Phys Org Chem 31:143
Denmark SC, Thorarensen A (1996) Chem Rev 96:137
Wade PA, Murray JK, Shah-Patel S, Caroll PJ (2002) Tetrahedron Lett 43:2585
Wade PA, Murray JK, Shah-Patel S, Le HT (2002) Chem Commun 1090
Sustmann R, Tappanchai S, Bandmann H (1996) J Am Chem Soc 118:12555
Kataoka F, Shimizu N, Nishida S (1980) J Am Chem Soc 102:711
Vassilikoginnakis G, Orfanopoullos M (1998) Tetrahedron Lett 39:8891
Jasiński R, Kwiatkowska M, Barański A (2009) J Mol Struct (TheoChem) 910:80
Arroyo P, Picher MT, Domingo LR (2004) J Mol Struct (TheoChem) 709:45
Arroyo P, Picher MT, Domingo LR, Terrier F (2005) Tetrahedron 61:7365
Domingo LR, Aurell MJ, Kneetman MN, Mancini PM (2008) J Mol Struct (TheoChem) 853:68
Steglenko DV, Kletsky ME, Kurbatov SV, Tatarov AV, Minkin VI, Goumont R, Terrier F (2009) J Phys Org Chem 22:298
TYa Paperno, Perekalin VV (1972) Infrakrasnye Spektry Nitrosoedinenii, Izd. GPI im. Gertsen’a, Leningrad
Streitwieser A, Jagow RH, Fahey RC, Suzuki S (1958) J Am Chem Soc 80:2326
O’Leary MH, Marlier JF (1978) J Am Chem Soc 100:2582
Jasiński R, Barański A (2010) Czasopismo Techn. PK (Chemia) (in press)
Ciosłowski J, Barański A, Juška T (1986) Tetrahedron 42:4549
Vogel AI, Furniss BS, Hannaford AJ, Smith PWG, Tatchell AR (1996) Textbook of practical organic chemistry, 5th edn. Wiley, New York
Barański A, Lyubimtsev A, Jasiński R, Kwiatkowska M (2008) Pol J Chem 82:1037
Kistiakowsky GB, Lacher JR (1936) J Org Chem 58:123
Schwietlick K (1971) Kinetische Metoden zur Untersuchung von Reaktionsmechanismen. VEB Deutscher Verlag der Wissenschaften, Berlin
Acknowledgments
Generous allocation of computing time by the regional computer center “Cyfronet” in Cracow (grant MNiI/SGI2800/PK/053/2003) and financial support from the Polish Ministry of Science and Higher Education (grant C2/263/DS/2008) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kwiatkowska, M., Jasiński, R., Mikulska, M. et al. Secondary α-deuterium kinetic isotope effects in [2+4] cycloaddition of (E)-2-phenylnitroethene to cyclopentadiene. Monatsh Chem 141, 545–548 (2010). https://doi.org/10.1007/s00706-010-0292-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-010-0292-6