
Abstract
The complete genome sequence of a new caulimovirus in Pueraria montana was determined using high-throughput sequencing. The 7,572 nucleotide genome of pueraria virus A (PVA) contains genes that encode a movement protein, an aphid transmission factor, a virion-associated protein, a coat protein, a protease + reverse transcriptase + ribonuclease H, and a transactivator/viroplasmin protein, as well as two intergenic regions, which are all common features of members of the genus Caulimovirus. A sequence alignment revealed that the complete genome of PVA shares 66.82% nucleotide sequence identity with strawberry vein banding virus (GenBank accession no. KX249738.1). The results of phylogenetic analysis and the observation that the nucleotide sequence of the polymerase coding region differed by more than 20% indicated that PVA is a member of a new species the genus Caulimovirus, family Caulimoviridae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The members of the family Caulimoviridae infect various plants worldwide. The family includes 11 genera, whose members have a monopartite, open, circular dsDNA genome of 7.1–9.8 kbp with discontinuities in both strands that is encapsidated in an icosahedral or bacilliform-shaped viral coat [1]. Like those of other viruses in the order Ortervirales, the genome of members of the family Caulimoviridae alternates between dsDNA and ssRNA through cycles of transcription and reverse transcription. However, in contrast to retroviruses, the dsDNA form of the viral genome is encapsidated rather than the ssRNA replication intermediate [2]. Caulimovirus genomes contain six to seven open reading frames (ORFs) that sequentially encode a movement protein (MP), an aphid transmission factor (ATF), a virion-associated protein, a coat protein (CP), a protease + reverse transcriptase (RT) + ribonuclease (RNase) H, and a transactivator/viroplasmin (TAV) protein, with two to four discontinuities in the strands [1].
Pueraria montana (commonly called kudzu) is one of 26 plant species of the genus Pueraria (family Fabaceae), which are mostly found in Asia, North America, and South America [3]. These plants, especially their tubers, are known for their traditional health and cosmetic benefits, as well as for their use in agriculture to prevent soil erosion [3]. Plants of the genus Pueraria are infected by various viral pathogens, including kudzu mosaic virus (genus Begomovirus), tobacco ringspot virus (genus Nepovirus), and soybean vein necrosis virus (genus Orthotospovirus) [4,5,6].
In this paper, we report the discovery of new caulimovirus in Pueraria montana (P. montana) and describe its complete genome sequence and organization.
P. montana leaf samples showing vein-clearing-like symptoms were collected from Chunyang-myeon, South Korea, in August 2018 (Fig. 1a) and were kept in powdered form at −80°C until used. To identify viral sequences, the collected P. montana samples and 16 other plant samples (n = 17) showing virus-like symptoms were pooled together as described previously [7,8,9]. A WizPrep™ Plant RNA Mini Kit (Seongnam, Korea) was used to extract total RNA from the pooled sample, and high-throughput paired-end RNA sequencing was performed after removing plant ribosomal RNA using a Ribo-Zero™ rRNA Removal Kit (Plant Leaf) (Epicentre, Madison, WI, USA). A cDNA library was constructed in accordance with the manufacturer’s instructions using a TruSeq RNA Sample Prep Kit (Illumina, San Diego, CA, USA). BluePippin™ 2% Agarose Gel Cassettes (Saga Science, Beverly, MA, USA) and an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA) were used to measure the size and quality of the cDNA, respectively. An Illumina NovaSeq6000 system was used to obtain paired-end reads. Approximately 82 GB of raw data were generated from the pooled samples. All of the raw reads were trimmed, and the subsequent de novo assembly and contig annotation were done by Macrogen (Seoul, Korea).

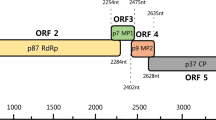
Disease symptoms and genome organization of a new caulimovirus identified in Pueraria montana. (A) Pueraria montana plant leaves with vein clearing symptoms. (B) Circular representation of the PVA genome. (C) Linear representation of the PVA genome. Conserved domains/motifs, such as the movement protein (MP), aphid transmission factor (ATF), virion-associated protein, peptidase, reverse transcriptase, viroplasmin, and the cysteine motif, were identified.
The resulting contig sequences were compared with available sequences in GenBank using a BLASTn search, which indicated that the test samples were infected with several known and unknown plant viruses. Among the contigs, one long caulimovirus-related contig (7,572 nucleotides [nt]), which was assembled from 2,445,773 reads, was identified. The contig shared the highest sequence identity – 66.82%, 65.89%, and 65.74% – with strawberry vein banding virus (KX249738.1), angelica bushy stunt virus (NC_043523.1), and cauliflower mosaic virus (AB863145.1), respectively, and it appeared to represent a novel caulimovirus. Accordingly, we considered this caulimovirus-related assembled sequence (7,572 nt) to be a complete genome sequence of a novel caulimovirus, which we tentatively designated as "pueraria virus A" (PVA).
To confirm the HTS result and test specifically for the virus in each plant sample, two primers, PVA_4457_F (TTGGCTTGAAACAAGCTCCT) and PVA _4895_R (TCCTGCTGTGTCCATATCCA) were designed based on the single caulimovirus-related contig sequence. Total DNA was extracted from each of the symptomatic 17 samples that were used for HTS, using a DNeasy® Plant Mini Kit (QIAGEN, Hilden, Germany). The extracted DNA samples were subjected to PCR using AccuPower® ProFi Taq PCR PreMix (Bioneer, Daejeon, Korea). Among the tested samples (n = 17), the sample from P. montana was the only one positive for PVA.
To confirm the complete genome sequence of PVA, seven additional primer sets were designed based on the PVA contig sequence (Supplementary Table S1). All of these primer sets were used successfully to amplify viral DNA from the P. montana sample (Fig. 1a). Using AccuPower® ProFi Taq PCR PreMix (Bioneer), PCR products of the expected sizes were obtained (Supplementary Fig. S2). All of the amplicons were purified using an AccuPrep PCR Purification Kit (Bioneer) and cloned independently into the RBC T&A Cloning Vector (RBC Bioscience, Taipei, Taiwan). To reduce experimental errors, at least three clonal inserts per PCR were sequenced using the Sanger method at Genotech (Daejeon, Korea). All of the overlapping PVA sequences were assembled using the DNAMAN 5.0 program (Lynnon Biosoft, Quebec, Canada). The assembled complete genome sequence of PVA shared a significant amount of sequence identity (65.2%) with other members of the genus Caulimovirus.
The complete 7,572-nt genome sequence of PVA was deposited in the GenBank database (accession no. MZ826138), and it shares the most sequence similarity (66.82% identity and 31% query coverage) with strawberry vein banding virus (GenBank accession no. KX249738.1).
The genome of PVA starts with the conserved tRNAMet sequence 5′-TGGTATCAGAGCC-3′, which is a primer binding site and complementary to the consensus sequence of the plant tRNAMet binding site [10]. It is therefore presumed to utilize host tRNA molecules as primers for genome replication by reverse transcription of the negative-sense DNA strand [11].
Six putative ORFs were identified in the complete PVA genome sequence (Fig. 1b and c) using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/): ORF1, nt 40–1,005; ORF2, nt 1,002–1,436; ORF3, nt 1,514–1,936; ORF4, nt 1,933–3,354; ORF5, nt 3,311–5,452; and ORF6, nt 5,566–6,876 (Fig. 1c). For the detection and functional annotation of the conserved domains/motifs in protein-encoding sequences, the Pfam database of protein families (http://pfam.xfam.org/) [12] was used (Fig. 1c).
ORF1 of PVA encodes an MP. The product of ORF1, which has two conserved amino acid sequence motifs, GNLSYGKLMF (aa 166–175) and GYTLSNSHHS (aa 220–229), is believed to be involved in cell-to-cell movement of caulimovirus members [8, 13, 14]. ORF2 of PVA encodes an IXG motif, which is necessary for interaction between the ATF and viral particles during aphid transmission in members of the genus Caulimovirus [8, 13, 14]. ORF3 of PVA potentially encodes a multifunctional virion-associated protein, which might play a role in virus cell-to-cell and plant-to-plant transmission, and interacts with the capsid, movement protein, and aphid transmission factor [15].
ORF4 of PVA encodes a viral CP, which contains a conserved cysteine motif (CX2CX4HX4C, aa 404–418). A similar motif has been identified in another caulimovirus CPs, and it includes an RNA-binding domain that is consistent with a cysteine motif or ‘zinc finger’ [16]. ORF5 of PVA encodes a polyprotein containing all of the motifs conserved in caulimovirus replicases, including aspartic protease (aa 54–213), reverse transcriptase (RT) (aa 334–489), and RNase H (aa 579–687) motifs, making it similar to the putative protease domains reported previously for caulimoviruses [8, 13, 17]. The conserved RT domain of caulimoviruses is present as YVDDIVF (aa 438–445) and IIETDASDLYWG (aa 484–495). ORF6 of PVA encodes a caulimovirus viroplasmin, with a conserved TAV (GLCSIIY; aa 250–256), which is critical for viral replication, translation, assembly, and protection against plant defense mechanisms [9, 13, 18].
PVA has two intergenic regions. The smaller one is 160 nt in length and is found between ORF5 and ORF 6, whereas the longer one is between ORF1 and ORF6. The former has a tentative TATA-like box, TATATATA (nt 5519–5526), and the latter contains a putative polyadenylation signal, AATAAAA (nt 7137–7143), downstream from its TATA-like box [9, 13, 17].
Sequence comparisons showed that the overall nucleotide sequence identity between PVA and others caulimovirus members ranged from 43.07% to 51.35% (Supplementary Table S2). Furthermore, pairwise alignments of amino acid sequences of PVA ORFs with those of another caulimoviruses revealed low sequence similarity (12.77%–47.90% identity). Only ORF5 had relatively high amino acid sequence similarity (62.46%–42.52% identity) to other members of the genus Caulimovirus (Supplementary Table S2). This suggests that ORF5 of PVA has a closer evolutionary relationship to other caulimoviruses than the other ORFs. To better understand the molecular relationships between PVA and other caulimoviruses, a phylogenetic tree was constructed by the maximum-likelihood method with 1000 bootstrap replicates in MEGA X v. 10.1.8 [19] using amino acid sequences. The phylogenetic tree constructed using an amino acid alignment of ORF 5 from PVA and other members of the genus Caulimovirus placed PVA within a group corresponding to the genus Caulimovirus and closest to SVBV (Fig. 2).

Phylogenetic tree based on amino acid sequences of ORF5 of PVA and other members of the genus Caulimovirus. The maximum-likelihood phylogenetic tree was constructed by the JTT matrix-based model in MEGA X v. 10.1.8, using ClustalW for sequence alignment. Bootstrap values for 1000 replicates > 50% are shown at the branches. Soybean chlorotic mottle virus (SbCMV) and blueberry red ringspot virus (BRRV) were used as the outgroup.
In summary, pueraria virus A (PVA) fulfills the current International Committee on Taxonomy of Viruses species demarcation criteria for caulimoviruses, which include host range differences and differences in polymerase (RT + RNase H) nt sequences of more than 20%. Thus, it can be classified as a member of a new species in the genus Caulimovirus. The genomic sequence obtained in this study will help in the further characterization of this virus and identification of other potential hosts. Since P. montana propagates vegetatively, our finding of another distinct caulimovirus highlights the importance of developing virus-specific detection and management alternative for these viruses. Further research is needed to identify the vector and to investigate the possible presence of plant-genome-integrated subgenomic forms or episomal elements.
References
Teycheney P-Y, Geering ADW, Dasgupta I et al (2020) ICTV virus taxonomy profile: caulimoviridae. J Gen Virol 101:1025–1026. https://doi.org/10.1099/jgv.0.001497
Mart K, Jonas B, Coffin MJ et al (2021) Ortervirales: new virus order unifying five families of reverse-transcribing viruses. J Virol 92:e00515-e518. https://doi.org/10.1128/JVI.00515-18
Wang S, Zhang S, Wang S et al (2020) A comprehensive review on Pueraria: insights on its chemistry and medicinal value. Biomed Pharmacother 131:110734. https://doi.org/10.1016/J.BIOPHA.2020.110734
Zhang J, Wu ZJ (2012) First Report of Kudzu mosaic virus on Pueraria montana (Kudzu) in China. Plant Dis 97:148. https://doi.org/10.1094/PDIS-07-12-0671-PDN
Khankhum S, Bollich P, Valverde RA (2013) First report of Tobacco ringspot virus infecting kudzu (Pueraria montana) in Louisiana. Plant Dis 97:561. https://doi.org/10.1094/PDIS-10-12-0933-PDN
Zhou J, Aboughanem-Sabanadzovic N, Sabanadzovic S, Tzanetakis IE (2018) First report of soybean vein necrosis virus infecting Kudzu (Pueraria montana) in the United States of America. Plant Dis 102:1674. https://doi.org/10.1094/PDIS-01-18-0042-PDN
Liu H, Zhao F, Qiao Q et al (2021) Complete genome sequence of a divergent sweet potato chlorotic stunt virus isolate infecting Calystegia hederacea in China. Arch Virol 166:2037–2040. https://doi.org/10.1007/s00705-021-05076-0
Lim S, Baek D, Igori D, Moon JS (2017) Complete genome sequence of a putative new caulimovirus which exists as endogenous pararetroviral sequences in Angelica dahurica. Arch Virol 162:3837–3842. https://doi.org/10.1007/s00705-017-3517-8
Lim S, Igori D, Zhao F et al (2015) Complete genome sequence of a tentative new caulimovirus from the medicinal plant Atractylodes macrocephala. Arch Virol 160:3127–3131. https://doi.org/10.1007/s00705-015-2576-y
Pooggin MM, Ryabova LA (2018) Ribosome shunting, polycistronic translation, and evasion of antiviral defenses in plant pararetroviruses and beyond. Front Microbiol 9:644. https://doi.org/10.3389/fmicb.2018.00644
Menéndez-Arias L, Sebastián-Martín A, Álvarez M (2017) Viral reverse transcriptases. Virus Res 234:153–176. https://doi.org/10.1016/j.virusres.2016.12.019
Finn RD, Bateman A, Clements J et al (2014) Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. https://doi.org/10.1093/nar/gkt1223
Petrzik K, Beneš V, Mráz I et al (1998) Strawberry vein banding virus—definitive member of the genus Caulimovirus. Virus Genes 16:303–305. https://doi.org/10.1023/A:1008039024963
Richins RD, Scholthof HB, Shepherd RJ (1987) Sequence of figwort mosaic virus DNA (caulimovirus group). Nucleic Acids Res. https://doi.org/10.1093/nar/15.20.8451
Leclerc D, Stavolone L, Meier E et al (2001) The product of ORF III in cauliflower mosaic virus interacts with the viral coat protein through its C-terminal proline rich domain. Virus Genes 22:159–165. https://doi.org/10.1023/A:1008121228637
Pahalawatta V, Druffel KL, Wyatt SD et al (2008) Genome structure and organization of a member of a novel and distinct species of the genus Caulimovirus associated with dahlia mosaic. Arch Virol 153:733–738. https://doi.org/10.1007/s00705-008-0043-8
Eid S, Almeyda CV, Saar DE et al (2011) Genomic characterization of pararetroviral sequences in wild Dahlia spp. in natural habitats. Arch Virol 156:2079. https://doi.org/10.1007/s00705-011-1076-y
Pappu HR, Druffel KL (2009) Use of conserved genomic regions and degenerate primers in a PCR-based assay for the detection of members of the genus Caulimovirus. J Virol Methods 157:102–104. https://doi.org/10.1016/J.JVIROMET.2008.11.014
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547
Acknowledgements
This work was supported by IPET (Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries; Project No. AGC1762111), Ministry of Agriculture, Food and Rural Affairs, Republic of Korea. Also, this study was supported by the Animal and Plant Quarantine Agency (No. FDM0012211) Republic of Korea. We thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies of involving human participants or animals.
Additional information
Handling Editor: Elvira Fiallo-Olivé.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gudeta, W.F., Igori, D., Belete, M. . et al. Complete genome sequence of pueraria virus A, a new member of the genus Caulimovirus. Arch Virol 167, 1481–1485 (2022). https://doi.org/10.1007/s00705-022-05431-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-022-05431-9