
Abstract
A novel positive-strand RNA virus infecting citrus with the tentative name “citrus leaf blotch virus 2” (CLBV-2), was identified in the present work. The complete genome sequence of CLBV-2 comprises 8,697 nucleotides (nt) excluding a poly(A) tail and three open reading frames (ORFs), showing the highest nucleotide sequence identity with the Actinidia strain (JN983456) of citrus leaf blotch virus (CLBV). The putative movement protein (ORF2), coat protein (ORF3), and 3′ untranslated region (UTR) shared high sequence similarity with those of the extant CLBV isolates. In contrast, only low sequence similarity was observed in the 5′ UTR and putative replicase polyprotein (ORF1) regions. The distant phylogenetic relationship between CLBV-2 and CLBV was deduced based on whole-genome nucleotide and whole-ORF1 amino acid sequence comparisons. Sequence comparisons suggest that CLBV-2 acquired an ORF2-ORF3-3′ UTR region homologous to CLBV by recombination with of an unknown citrivirus. In view of the fact that this genomic recombination event appears to have occurred between members of different species in the genus Citrivirus, we propose that CLBV-2 should be considered a member of a distinct species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Citrus leaf blotch virus (CLBV) is the only current member of the genus Citrivirus in the family Betaflexiviridae (https://talk.ictvonline.org/). The virus has a linear, positive-sense, single-stranded genomic RNA of approximately 8.7 kb that contains three open reading frames (ORFs). ORF1 encodes replicase polyprotein (RP) with conserved motifs for methyltransferase (Met), AlkB-like oxygenase (AlkB-Oxy), ovarian tumor-like protease (OTu-Pro), papain-like protease (P-Pro), helicase (Hel) and RNA-dependent RNA polymerase (RdRp). ORF2 encodes a putative movement protein (MP), which acts as silencing suppressor [9]. ORF3 encodes a coat protein (CP) [10].
CLBV infects various hosts, including fruit-bearing, ornamental and herbaceous plants [1,2,3, 12]. Interestingly, probably due to viral dispersal through grafting and seeding, isolates from various citrus types and geographical areas show a stable genetic structure [5, 11]. Recently, however, we discovered a novel isolate from the citrus variety Haruka (C. tamuranua) using next-generation sequencing (NGS). This isolate showed marked genetic divergence from the known isolates, suggesting that it represents a different type of citrivirus.
Haruka is a natural variant of Hyuganatsu, which is a hybrid of yuzu (C. junos) and pomelo (C. grandis). It is a pedigreed and excellent cultivar introduced from Japan that is currently popular in China. Its planting area in Xiangshan county, Zhejiang, covered 223 hm2 in 2015 with annual earnings of 5,000 U.S. dollars per acre. One Haruka tree exhibiting leaf chlorotic blotching (Fig. 1a), collected from a field in Chongqing, was preserved in an isolation chamber, and its leaves were stored at –80°C. Total RNA was extracted using TRIzol Reagent (Invitrogen) according to the manufacturer’s specifications and sent to a sequencing company (Biomarker) for analysis by Illumina next-generation sequencing (NGS) (Table S1). Standard data analysis, including processing raw reads as well as de novo assembly of clean reads (Table S1) and BLAST searches of assembled contigs in NCBI databases resulted in eight viral contigs: seven matched CLBV sequences, and one was related to citrus tristeza virus (CTV). All eight contigs were confirmed by PCR assays, and only the CLBV-related contigs were used for further study.

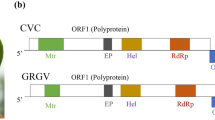
(a) Chlorotic blotching of leaves associated with CLBV-2 infection in Haruka. (b) Genome organization of CLBV-2. (c) Nucleotide and amino acid pairwise identity between CLBV-2 and representative members of the family Betaflexiviridae in the whole genome, coding-region, and noncoding-region. The deduced proteins and protein domains are shown as boxes. The lighter the blue or deeper the orange, the higher the identity (na: not applicable)
To reconstruct the viral genome, eight pairs of specific primers (Table S2) were initially designed based on the viral contigs. A two-step RT-PCR was carried out using a reverse transcription (RT) kit (Promega) and a PCR kit (Takara). The 5′- and 3′-terminal sequences of the genome were determined using a commercial RACE kit (Invitrogen). Amplicons of the expected size were purified using an Agarose Gel DNA Extraction Kit (OMEGA Biotek) and ligated into a pEASY-T1 Cloning Vector (TransGen Biotech). At least five clones per amplicon were completely sequenced, and sequences were assembled using DNASTAR7 (DNASTAR Inc.). The genomic ORFs were predicted using ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/), and conserved protein domains were identified using the Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Sequence analysis was conducted using CLC Genomics Workbench 9.5, and phylogenetic analysis was performed in MEGA 7 using the maximum-likelihood algorithm with 1000 bootstrap replicates [4].
The complete genome sequences of the virus, which was given the name “citrus leaf blotch virus 2” (CLBV-2) was 8,697 nt in length, excluding the poly(A) tail, and it has been deposited in the GenBank database with the accession number MH144344. At the whole-genome level, CLBV-2 was found to be 63.2–64.3% identical to all 13 CLBV isolates whose sequences were retrieved from GenBank, including Citrus isolates (AJ318061, EU857539-40, FJ009367, MF784853-56); Actinidia isolates (JN900477, JN983454-56); and a Prunus isolate (KR023647). The genomic organization was similar to that of CLBV, containing three ORFs (Fig. 1b). Interestingly, the full-length genome was 50, 65 and 85 nt shorter than those of the Citrus, Prunus, and Actinidia isolates, respectively. In addition, the genomic 5′-termini began with CGAAAA, which differed slightly from the GAAAA found at the 5’ ends of CLBV isolates. The 5′ untranslated region (UTR) was 71 in length, sharing 44.6–57.5% identity with those of the CLBV isolates, with which the 3′ UTR (526 nt) showed 67.6–86.4% identity.
The large ORF1 reached from AUG at nt position 72 to an “opal” termination codon (UAG) at nt 5,927 and was predicted to encode a 1,951-aa RP with an estimated molecular mass of approximately 228.08 kDa. As is typical for citriviruses, the RP had six conserved domains: Met (aa 44–341), AlkB-Oxy (aa 840–947), Hel (aa 1,153–1,399), RdRp (aa 1,569–1,899), OTu-Pro (aa 676–799) and P-Pro (aa 969–1,055; Cys973 as a potential catalytic site) (Fig. 1b) [7]. However, the RP was 11 aa shorter than that of the Citrus isolates, mainly due to a 24-aa deletion between aa positions 120 and 143. Additionally, it shared only 56.6–57.2% nucleotide and 51.9–52.4% amino acid identity with CLBV isolates. ORF2 (nt 5,928 to 7,016) was predicted to encode a 362-aa MP with a calculated molecular weight of approximately 40.4 kDa, with high nucleotide (76.2–78.6%) and amino acid sequence identity (89.8–91.7%) to those of CLBV isolates. ORF1 and ORF2 in CLBV were separated rather than overlapping. Between ORF2 and ORF3, there was a 63-nt intercalated non-coding region. The 363-aa protein encoded by ORF3 (nt 7,080 to 8,171) was predicted to function as a CP. Its predicted size was approximately 40.62 kDa, sharing 76.4–85.6% nucleotide and 87.6–95.1% amino acid sequence identity with those of CLBV isolates.
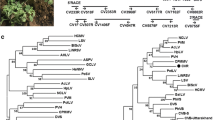
A phylogenetic tree based on the genome sequence (Fig. 2) and the RP gene sequence (Fig. S1a) of CLBV-2 and representative members of the family Betaflexiviridae clearly showed that CLBV-2, although it clustered with CLBV, was placed in a distinct clade. Nevertheless, CLBV-2 grouped with CLBV in a tree constructed based on the CP gene sequence (Fig. S1b).

Evolutionary relationships of CLBV-2 with representatives of different genera and an unassigned species in the family Betaflexiviridae, inferred from whole-genome sequence comparisons. MEGA 7.0 was used to perform a MUSCLE alignment, and the maximum-likelihood algorithm (Jukes-Cantor model) with 1000 bootstrap replicates was used to construct the tree. Bootstrap percent identity values less than 50 are not shown. Accession numbers for each virus are shown near the virus name
In the family Betaflexiviridae, genomic recombination plays very important role in virus evolution [6]. Therefore the genome sequences of the CLBV-2 and 13 CLBV isolates were analyzed using RDP4 [8] software to find evidence of recombination. Five probable recombination events of in the CLBV-2 genome were predicted, with an unknown virus as the major parent at nt 1 to 5,637 (the 5′ UTR and RP gene) and some CLBV isolates as minor parents successively at nt 5,638 to 8,697 (the MP, CP genes and 3′ UTR) (Table S3 and Fig. S2).
According to the species demarcation criteria established by the International Committee on Taxonomy of Viruses (ICTV) for the family Betaflexiviridae (less than 72% nucleotide or 80% amino acid sequence identity in the CP or RP gene), CLBV-2 should be considered a member of novel citrivirus species, as indicated by sequence comparisons between the RP gene (Fig. 1c). On the other hand, this virus appears to have originated through RNA recombination between members of two distinct citrivirus species, resulting in the MP and CP genes being clearly homologous to those of CLBV (Fig. 1c).
Recently, a new CLBV-2 variant, CN2 (8,698 nt; MH558590), with a genome sequence and symptoms similar to those of the CLBV-2 isolate (92% identity) was obtained from a Hyuganatsu tree showing no CTV infection, in an orchard in Chongqing, suggesting that this type of recombinant is stable under natural selective pressure. We therefore conclude that CLBV-2 represents a new species within the genus Citrivirus. So far, CLBV-2 has only been detected in two citrus trees. Whether it originated from or infects other trees has not been determined, but the fact that its spread has not been observed suggests that viral transmission is not favored under natural conditions.
Change history
22 October 2018
Unfortunately, the Acknowledgement, ethical statement and the Conflict of interest statements were not included in the online publication and updated here in this Erratum.
References
Chavan RR, Blouin AG, Cohen D, Pearson MN (2013) Characterization of the complete genome of a novel Citrivirus infecting Actinidia chinensis. Arch Virol 158:1679–1686
Galipienso L, Vives MC, Moreno P, Milne RG, Navarro L, Guerri J (2001) Partial characterisation of Citrus leaf blotch virus, a new virus from Nagami kumquat. Arch Virol 146:357–368
Gress JC, Smith S, Tzanetakis IE (2016) First report of Citrus leaf blotch virus in Peony in the USA. Plant Dis 101:637
Kumar S, Nei M, Dudley J, Tamura K (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform 9:299–306
Li P, Li M, Zhang S, Wang J, Yang FY, Cao MJ, Li ZA (2018) Complete genome sequences of four isolates of Citrus leaf blotch virus from citrus in China. J Integr Agric 17:712–715
Marais Faure, Mustafayev Candresse (2015) Characterization of new isolates of and of a new: evidence for recombination as a driving force in evolution. Plos One 10:e0129469
Martelli GP, Adams MJ, Kreuze JF, Dolja VV (2007) Family Flexiviridae: a case study in virion and genome plasticity. Annu Rev Phytopathol 45:73
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003
Renovell Á, Vives MC, Ruiz-Ruiz S, Navarro L, Moreno P, Guerri J (2012) The Citrus leaf blotch virus movement protein acts as silencing suppressor. Virus Genes 44:131–140
Vives M, Galipienso L, Navarro L, Moreno P, Guerri J (2001) The nucleotide sequence and genomic organization of Citrus leaf blotch virus: candidate type species for a new virus genus. Virology 287:225–233
Vives MC, Rubio L, Galipienso L, Navarro L, Moreno P, Guerri J (2002) Low genetic variation between isolates of Citrus leaf blotch virus from different host species and of different geographical origins. J Gen Virol 83:2587
Wang J, Zhu D, Tan Y, Zong X, Wei H, Liu Q (2016) First report of Citrus leaf blotch virus in sweet cherry. Plant Dis 100:1027
Author information
Authors and Affiliations
Corresponding authors
Additional information
Handling Editor: Stephen John Wylie.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Phylogenetic maximum-likelihood trees derived from RP (a) and CP (b) amino acid sequence alignments (MUSCLE) of CLBV-2 with selected viruses of the family Betaflexiviridae, using the JTT model and 1000 bootstrap replicates (values less than 50% are not shown). Each virus is indicated by a full name and an accession number (JPEG 364 kb)
Fig. S2
Two different types of recombination of CLBV-2 with the same major parent (unknown) but different minor parents: (a) CLBV-Citrus and -Prunus (with four recombinants) and (b) CLBV-Actinidia (with one recombinant) (JPEG 238 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Cao, M., Li, P., Zhang, S. et al. Molecular characterization of a novel citrivirus from citrus using next-generation sequencing. Arch Virol 163, 3479–3482 (2018). https://doi.org/10.1007/s00705-018-4039-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-4039-8