
Abstract
The emerged influenza A/H1N1pdm09 viruses have replaced the previously circulating seasonal H1N1 viruses. The close antigenic properties of these viruses to the 1918 H1N1 pandemic viruses and their post-pandemic evolution pattern could further enhance their adaptation and pathogenicity in humans representing a major public health threat. Given that data on the dynamics and evolution of these viruses in Saudi Arabia is sparse we investigated the genetic diversity of circulating influenza A/H1N1pdm09 viruses from Jeddah, Saudi Arabia, by analyzing 39 full genomes from isolates obtained between 2014-2015, from patients with varying symptoms. Phylogenetic analysis of all gene segments and concatenated genomes showed similar topologies and co-circulation of clades 6b, 6b.1 and 6b.2, with clade 6b.1 being the most predominate since 2015. Most viruses were more closely related to the vaccine strain (Michigan/45/2015) recommended for the 2017/2018 season, than to the California/07/2009 strain. Low sequence variability was observed in the haemagglutinin protein compared to the neuraminidase protein. Resistance to neuraminidase inhibitors was limited as only one isolate had the H275Y substitution. Interestingly, two isolates had short PA-X proteins of 206 amino acids compared to the 232 amino acid protein found in most influenza A/H1N1pdm09 viruses. Together, the co-circulation of several clades and the predominance of clade 6b.1, despite its low circulation in Asia in 2015, suggests multiple introductions most probably during the mass gathering events of Hajj and Umrah. Jeddah represents the main port of entry to the holy cities of Makkah and Al-Madinah, emphasizing the need for vigilant surveillance in the kingdom.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Influenza viruses are contagious respiratory pathogens and one of the most common causes of acute respiratory infections in humans worldwide. They are associated with periodic outbreaks and epidemics as well as occasional pandemics causing substantial health and economic burdens [1]. Annual influenza epidemics affect 5 to 15% of the world’s population and cause severe disease and hospitalizations in up to 5 million people as well as up to 500,000 deaths globally [2, 3]. Severe disease and death are most pronounced in high-risk groups; the elderly (> 65 years of age), young children (< 5 years of age), pregnant women, immunocompromised patients and individuals with chronic diseases [3, 4].
Among all influenza viruses, influenza A viruses (IAVs) are the most diverse with a wide host range and multiple subtypes which comprise different combinations of the 18 hemagglutinins (HAs) and 11 neuraminidases (NAs) known to date [5]. They undergo a high rate of antigenic variations due to the lack of proofreading in the viral RNA-dependent RNA polymerase and the segmented nature of the genome. Specifically, antigenic drift results from an accumulation of point mutations which occurs in all IAV genes, but is most prominent in antigenic sites targeted by neutralizing antibodies on the HA and NA surface glycoproteins. This usually leads to phenotypic changes affecting tropism, virulence, replication and antigenicity, ultimately causing annual epidemics [6, 7]. The second antigenic change is characterized by a major antigenic shift due to reassortment of viral segments during co-infections resulting in a new genome and new antigenic properties and leading to possible global pandemics, as seen in the last century [8, 9].
The first influenza pandemic of this century was announced in June 2009, with the emergence of the novel H1N1 IAV strain (A/H1N1pdm09) [10]. This triple reassortment H1N1 virus showed a rare reassortment event involving several gene segments of human, swine and avian origin. This included HA, nucleoprotein (NP) and non-structural (NS) protein genes from a classical swine influenza lineage, NA and matrix (M) protein genes derived from an Eurasian swine influenza lineage, polymerase segments (PB2 and PA) from the avian origin North American swine triple reassortment influenza lineage, and polymerase segment PB1 from a human origin swine triple reassortment influenza lineage [10,11,12,13]. Since 2010, A/H1N1pdm09 has replaced the previously circulating seasonal H1N1 strains and became endemic worldwide. Several studies have shown that influenza A/H1N1pdm09 is antigenically close to the devastating 1918 H1N1 pandemic and that its post-pandemic evolution has further enhanced its adaptation and pathogenicity in humans, raising several public health concerns [14,15,16,17,18,19].
Influenza antivirals are generally divided into two classes; ion channel (M2) blockers (adamantanes), including amantadine and rimantadine and NA inhibitors (NAIs) including oseltamivir, zanamivir, peramivir and laninamivir. Emergence of resistance to adamantanes since 2005/2006 made them ineffective and obsolete [20]. Therefore, NAIs, especially oseltamivir, are the drugs of choice for treatment and prophylaxis. However, resistance can always emerge with or without treatment. Therefore, routine surveillance for oseltamivir resistance is required to ensure the effectiveness of oseltamivir against circulating viruses. Fortunately, since the emergence of A/H1N1pdm09 in 2009, oseltamivir resistance is generally low. However, continuous surveillance and global monitoring of influenza viruses are critical to track vaccine-escape and antiviral resistant strains, monitor viral evolution, aid in the selection of the annual vaccine strains, and detect key amino acid changes involved in increased viral transmissibility and pathogenicity.
Unfortunately, the number of complete IAVs genome studies from Middle Eastern countries is relatively small. Analysis of the publicly available influenza databases clearly shows poor influenza surveillance in countries like Saudi Arabia, which receives millions of visitors annually for religious or work purposes. Introduction and transmission of influenza viruses as well as other respiratory pathogens is very common in mass gatherings such as the Hajj and Umrah pilgrimages in the holy cities of Makkah and Al-Madinah. During these religious events, Muslims from all over the world visit these cities, mostly through the city of Jeddah, and spend long times in very crowded areas posing the risk of introducing new viruses, not only to Saudi Arabia but also to their home countries [21,22,23]. Therefore, in this study we attempted to study the genetic diversity and evolution of influenza A/H1N1pdm09 viruses circulating in Jeddah, Saudi Arabia from late 2013-2014 to early 2015-2016.
Materials and methods
Samples
Nasopharyngeal (NP) swabs were collected over 3 influenza seasons: the late 2013/2014 season from April 2014 to June 2014, the 2014/2015 season from December 2014 to April 2015, and the early 2015/2016 season from October 2015 to November 2015. NP swabs were collected from all symptomatic patients with respiratory manifestations presenting at 2 tertiary hospitals in Jeddah, Saudi Arabia and screened for influenza A, B and A/H1N1 viruses as indicated below. Ethical approval was obtained from the Unit of Biomedical Ethics in King Abdulaziz University Hospital and the Research Ethics Board in King Fahad Armed Forces Hospital, Jeddah, Saudi Arabia.
Real-time reverse transcription PCR (rRT-PCR)
Viral RNA was extracted from all NP samples using the QIAamp Viral RNA mini kit according to the manufacturer’s instructions (Qiagen, USA) and stored at -80 °C until use. Extracted RNA from all samples was screened for influenza A, B and A/H1N1 viruses using real-time RT-PCR as previously described [24]. Only influenza A/H1N1 positive samples with cycle threshold (ct) values of < 30 (39 samples met the criteria, as shown in Table 1) were used for full genome sequencing at the Special Infectious Agents Unit (SIAU), King Fahd Medical Research Center (KFMDR), King Abdulaziz University (KAU).
Whole genome sequencing
Extracted RNA from the selected samples was used to amplify the 8 viral segments using sequencing primers and protocols as previously described [25]. RT-PCR products were gel-extracted, purified and used for Sanger sequencing on an ABI 3500 Automatic Sequencer (Applied Biosystems, Foster City, CA) using M13-forward and reverse primers and the Bigdye Terminator V3.1 Reaction Cycle Kit (Applied Biosystems) as previously described [26]. All obtained sequences were deposited in the NCBI GenBank database under accession numbers MF768576 to MF768887.
Phylogenetic analysis
Obtained sequences were multiply aligned by MUSCLE [27], and edited in Geneious Software version 7.1.9 5.0.9 [28]. Sequences from this study, recently reported Saudi isolates from 2013 to 2016, as well as reference strains representing the different clades of A/H1N1pdm09 including A/H1N1pdm09 vaccine strains (California/07/2009 and Michigan/45/2015) were retrieved from the NCBI Influenza Resource Database (http://www.ncbi.nlm.nih.gov/genomes/FLU) and the Global Initiative on Sharing All Influenza Data (GISAID; http://platform.gisaid.org/) databases and used for phylogenetic tree construction using the MEGA 6.0 software [29]. Concatenated sequences were aligned, manually inspected, trimmed and concatenated to generate data sets, each of 13,133 bp in length and in the following sequence order: PB2, PB1, PA, HA, NP, NA, M and NS (using Geneious Software). The evolutionary history was inferred by the Maximum Likelihood (ML) method based on a best-fit nucleotide substitution model for each segment as implemented in MEGA6 with a bootstrap analysis of 1,000 replicates (75% support threshold). The Tamura 3-parameter model (+G) was used for the HA and NA genes [30], the Kimura 2-parameter model for the M gene, the Kimura 2-parameter model (+I) for the NS gene [31], the Hasegawa-Kishino-Yano model for NP, PA, PB1 and PB2 [32], and discrete Gamma distribution (+G+I) [33] was used for concatenated sequences. Initial trees were obtained by applying the Neighbor-Joining method using the Maximum Composite Likelihood (MCL) approach. The trees are drawn to scale, and branch lengths were measured in the number of substitutions per site.
Results
Predominance of influenza A/H1N1pdm09 clade 6b.1 from early 2015 onwards
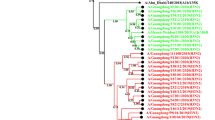
Influenza A/H1N1pdm09 viruses have evolved into eight major genetic clades (1 to 8) in which clade 6 has further diverged into 6A, 6B, 6C, 6B.1, and 6B.2 based on HA gene variation. Phylogenetic analysis of concatenated genomes of the 39 influenza A/H1N1pdm09 isolates obtained in 2014 and 2015 in this study and 5 previously reported Saudi strains from 2015 (Fig. 1) showed that all viruses are clustered forming monophyletic clades. All 2014/2015 Saudi isolates were from clade 6b represented by the South Africa/3626/2013 strain, in contrast to the 2013 Saudi isolates which clustered within clade 6c (Fig. 1). A phylogenetic tree of the HA genes (Fig. 2) further confirmed this finding and showed that 2014 and 2015 Saudi isolates are more closely related to the vaccine strain (Michigan/45/2015) recommended for the 2017/2018 season than the California/07/2009 strain which was used from 2009 until the 2016/2017 season. HA proteins from all Saudi 2014/2015 strains were characterized by previously known, clade 6b-signature amino acid substitutions in HA1 and HA2 (Table 2). However, most of the 2015 isolates (27 from the current study and 9 from previous reports) further clustered with Michigan/45/2015 in clade 6b.1 with additional distinguishing amino acid changes compared to the California/07/2009 vaccine strain, including S84N, S162N and I216T in HA1, and A13T in the signal peptide (Fig. 2). On the other hand, all isolates from 2014 (7 isolates) and 8 isolates from 2015 (4 from the current study and 4 from previous reports) lacked these mutations and fell into clade 6b. Only one Saudi 2015 strain (Jeddah/KFAFH1334/2015) clustered in clade 6b.2, represented by Massachusetts/01/2016 and had known clade 6b.2 amino acid substitutions (Fig. 2). Together, these results show that, since 2015, clade 6b.1 has been the most predominate IAV in Jeddah, Saudi Arabia.

Phylogenetic analysis of concatenated influenza A/H1N1pdm09 genomes. The 2014 and 2015 isolates from Jeddah characterized in this study are marked with closed and open circles, respectively. Reference influenza A/H1N1pdm09 viruses from the different clades are color coded

Phylogenetic analysis of influenza HA and NA genes. The trees with the highest log likelihood (-4325.3596) for both the HA and (-3545.2999) NA genes are shown. The analysis involved 79 nucleotide sequences with a total of 1701 and 1410 positions for HA and NA, respectively. Isolates with the H274Y mutation are highlighted with yellow in the NA tree. The 2014 and 2015 isolates from Jeddah characterized in this study are marked with closed and open circles, respectively. Reference vaccine strains are shown in green. Significant and signature amino acid substitutions are depicted on the tree. Substitutions shown in rectangles represent reversions
Low diversity in the HA of influenza A/H1N1pdm09 clade 6b.1 viruses circulating in Jeddah in 2015
Detailed examination of the HA proteins showed few variants with unique amino acid changes, e.g. only two mutations (H296N and K308R in HA1) were observed which have not been reported in influenza A/H1N1pdm09 viruses before (Supplementary Table). Interestingly, some of the isolates, such as Jeddah/KFAFH1568/2015 and Jeddah/9881/2015, showed reversions at position 13 in the signal peptide (T13A), while Jeddah/KFAFH1584/2015 had the V321I reversion mutation which is a characteristic marker for all clades, when compared to clade 1 viruses (Fig. 2). Two isolates, Jeddah/7611/2014 and Saudi Arabia/78085/2015, had D222N in the Ca antigenic site and N129D, respectively. Both of these substitutions have been suggested to be associated with increased disease severity [34,35,36]. Moreover, one variant (Jeddah/KFAFH3161/2015) showed a S190R substitution at the antigenic site Sb (Supplementary Table). Of note two Saudi isolates (Saudi Arabia/02/2013 and Saudi Arabia/03/2013) reported previously, clustered together in clade 6c with several shared mutations including A186T and R205K in antigenic sites Sb and Ca, respectively (Fig. 2). Nonetheless, our analysis showed low sequence variability in the HA protein of A/H1N1pdm09 isolates classified within clade 6b.1 (Supplementary Table).
High diversity, with low prevalence of NAI resistance-associated mutations, in influenza A/H1N1pdm09 viruses circulating in Jeddah in 2015
The overall topology in the NA tree was similar to that observed for HA (Fig. 2) in which all 2014/2015 Saudi strains fell into clade 6b with distinctive clade 6b-specific amino acid changes (Table 2). While the V106I substitution was seen as an early marker for isolates from most clades, compared to California/07/2009, clade 6b and 6c viruses gained a reversion at this position from isoleucine to valine. Similar to the HA tree, all isolates in clade 6b.1 clustered together with Michigan/45/2015 by acquiring further mutations (V13I, L40I, V264I, N270K, I314M and N386K) and this included NA from two isolates (Jeddah/TAM/2015 and Jeddah/9127/2015) that originally clustered in clade 6b in the HA tree. On the other hand, strains from clade 6b.2 clustered together with two signature substitutions, with the remaining 2014/2015 strains falling into clade 6b as observed in the HA tree (Fig. 2).
Several 2015 strains within clade 6b.1 showed reverse mutations such as K270N, S44N and D248N. Fourteen of the 2015 isolates from this study had the K270N reversion. While a higher rate of polymorphisms was observed in the NA protein compared to HA, most mutations were not fixed as definitive markers (Supplementary Table). Nonetheless, some variants gained substitutions at antigenic positions such as I396V and D451G or potential N glycosylation sites such as N386S (Fig. 2 and Supplementary Table). Saudi Arabia/02/2013 and Saudi Arabia/03/2013 strains grouped together in clade 6c as seen in the HA tree, with two unique mutations. Importantly, none of the isolates showed any mutation associated with resistance to NAIs except for one isolate (Jeddah/0252/2015) with the H275Y mutation, suggesting a low prevalence of NAI-resistant strains in Jeddah, regardless of the use of oseltamivir. Further, five other isolates including 2 from this study (Jeddah/AMH/2015 and Jeddah/0141/2015) and 3 previously reported strains (Saudi Arabia/12/2015, Saudi Arabia/13/2015, Saudi Arabia/78558/2015) had the I117M mutation in the viral NA, which has been suggested to be associated with some oseltamivir resistance.
Signature mutations in the internal proteins of Saudi Arabian influenza isolates
All other phylogenetic trees for the other gene segments showed similar topology, in which all 2014/2015 Saudi strains were defined as clade 6b (Figs. 3 and 4) with signature amino acid changes (shown in Table 2). Clade 6b.1 isolates further grouped together with the vaccine strain (Michigan/45/2015) with additional characteristic amino acid changes (Figs. 3 and 4). Specifically, several fixed mutations were observed in most clade 6b.1 isolates; namely M1 (Q208K), NS1 (E125D and D2E), NS2 (M83I), NP (A22T and M105T), PA (R361K), PA-X (N204S) and PB2 (R299K and S543T). However, clade 6b.2 isolates did not show any unique markers in most trees, as shown in Figs 3 and 4. Not surprisingly, all isolates in this study had the S31N mutation which is associated with resistance to adamantanes as well as a nonfunctional PB1-F2 protein, similar to all previously isolated A/H1N1pdm09 strains. In the NS gene tree (Fig. 3), a Saudi Arabia/11/2015 strain obtained in another study [21], from an Australian pilgrim during the 2015 Hajj season, was more closely related to 2015 Saudi isolates in clade 6b. Similarly, PB2 from Jeddah/KFAFH0089/2015 and Jeddah/0418/2015 isolates, in addition to the Michigan/45/2015 reference vaccine strain, fell within clade 6b, together with isolates from clade 6b.2 sharing one unique substitution (A184T) (Fig. 4), and suggesting possible intra-subtype reassortment of these gene segments.

Phylogenetic analysis of influenza M, NS and NP genes. The trees with the highest log likelihood (-1934.5611) for the M, (-1816.7815) NS, and (-3350.1972) NP genes are shown. The analysis involved 71 nucleotide sequences with a total of 982, 838 and 1497 positions for M, NS and NP, respectively. The 2014 and 2015 isolates from Jeddah characterized in this study are marked with closed and open circles, respectively. Reference vaccine strains are shown in green. Significant and signature amino acid substitutions are depicted on the tree

Phylogenetic analysis of influenza PA, PB1 and PB2 genes. The trees with the highest log likelihood (-4664.5321) for the PA, (-4750.2012) PB1, and (-5124.2633) PB2 genes are shown. The analysis involved 71 nucleotide sequences with a total of 2151, 2274 and 2280 positions for PA, PB1 and PB2, respectively. Isolates with a truncated PA coding region are highlighted in yellow in the PA tree. The 2014 and 2015 isolates from Jeddah characterized in this study are marked with closed and open circles, respectively. Reference vaccine strains are shown in green. Significant and signature amino acid substitutions are depicted on the tree. Substitutions shown in rectangles represent reversions
Interestingly, two isolates (Jeddah/9127/2015 and Jeddah/TAM/2015) had a nucleotide mutation at position 621 from T to A which resulted in a premature stop codon at 620-622, truncating the PA-X protein at 206 amino acids, instead of the longer version observed in all influenza A/H1N1pdm09 isolates (232 amino acids). Repeated sequencing of these two isolates (Fig. 5) further confirmed this observation. Notably, these two isolates seem to cluster together in all trees with minor nucleotide differences and 1-2 amino acids changes in the NA, PB1 and PB2 proteins (Supplementary Table).

Premature stop-codons in the PA-X coding region. Nucleotide and amino acid sequences from influenza A Jeddah/KFAFH1851/2015, as well as the two isolates with the truncated PA-X proteins (Jeddah/9127/2015 and Jeddah/TAM/2015) are shown. The premature stop codon is indicated
Discussion
During the 2009 pandemic and in the following 2009–2010 winter season, circulating influenza A/H1N1pdm09 viruses, closely related to the vaccine strain A/California/07/2009, were characterized by few amino acid substitutions. During these seasons, HA clade 3 viruses and variants from HA clade 4 emerged and became more prevalent. In the 2010–2011 season, influenza A/H1N1pdm09 viruses diverged further and formed multiple clades with several genetic markers. Clades 2, 3, 4, 5, 6, 7 were circulating globally in this season, and by 2011-2012, only a few influenza A/H1N1pdm09 viruses from clades 5 and 7 were circulating and clade 6a had started to emerge. Similarly, clade 7 and clade 6a were less prevalent during the 2012–2013 season, compared to the emerging clade 6b and 6c viruses which eventually replaced viruses from clade 6b in 2013–2014 [37]. Subsequently, clade 6b viruses diverged into 6b.1 and 6b.2 with the successful introduction of new characteristic changes by the 2015-2016 season. Both clades were predominant in Europe, North America and Oceania but not in Asia, Africa or South America by September 2015, and it was not until 2016 that clade 6b.1 became predominant globally [38,39,40]. Interestingly, in the current study we found that 6b.1 and 6b.2 were circulating in Jeddah, Saudi Arabia as early as October 2015 (Table 1) with viruses from clade 6b.1 being the most prevalent, regardless of its low circulation in the region. Both of these two clades reacted poorly with post-immunization sera from people immunized with vaccine containing influenza A/California/7/2009-like-strain [41, 42], and it was not until September 2016 a vaccine strain from clade 6b.1 (A/Michigan/45/2015 H1N1pdm09-like virus) was recommended for vaccine production which could explain the limited efficacy of influenza vaccines in recent years.
Our results also identified several unique mutations in most gene segments when compared to the A/California/04/2009 vaccine strain (Supplementary Table). As expected, all isolates harbored the S31N mutation in their M2 genes rendering them resistant to adamantanes. In addition, only one isolate (Jeddah/0252/2015) had the H275Y mutation in the NA protein consistent with the previously reported low prevalence of NAI-resistant strains in Saudi Arabia [24, 43]. The observed I117M mutations in the NA of 5 isolates in the current study has been proposed to be associated with oseltamivir resistance [44, 45]. However, it was subsequently shown that I117V but not I117M could reduce the sensitivity of A/H1N1pdm09 viruses to oseltamivir, with additional synergistic effects becoming apparent when combined with H275Y [46]. The D222N mutation, within the receptor-binding site in HA, has been proposed to alter HA receptor specificity by increasing binding to both human and avian receptors [47, 48], and is associated with severe and fatal influenza cases [34, 35]. Interestingly, we identified one isolate (Jeddah/7611/2014) with this mutation in a mild case which did not require hospitalization (Table 1) suggesting that this mutation might not be associated with severity [47, 49]. Several reversion mutations at distinctive positions within the A/H1N1pdm09 viruses were also seen is this study, especially in the NA and PB2 proteins, however, the functional and antigenic importance of these changes is unknown and this requires further study, especially if these reversions become fixed in upcoming seasons.
PA-X is a recently recognized protein encoded in a + 1 frame-shift open reading frame (X-ORF) in the PA segment [50, 51]. All A/H1N1pdm09 viruses are characterized by a truncated version of PA-X which comprises 191 amino acids of the PA N-terminal region and a 41 amino acid C-terminal X domain, compared to the full-length protein which has a 61 amino acid C-terminal region [52]. Generally, PA-X proteins have been suggested as virulence factors and immune modulators involved in the suppression of host antiviral responses together with other proteins such as NS1, PA, PB1, PB2 and PB1-F2 [53, 54]. Studies have shown that the truncated PA-X has reduced nuclease activity, compared to the full-length protein, which could render viruses less efficient in terms of replication and of reduced virulence in mice [55,56,57]. Interestingly, such truncations have also been shown to enhance adaptation, pathogenicity and transmissibility in pigs [58]. Therefore, it not clear whether finding isolates with even shorter PA-X proteins, as was demonstrated for two isolates obtained in this study (Fig. 4) could contribute to further human adaptation and/or compensate for other proteins such as the non-functional PB1-F2, which is found in all influenza A/H1N1pdm09 viruses [59].
Each year, 2-3 million people attend the Hajj pilgrimage, in addition to more than 6 million foreign and 12 million local visitors who attend the all year round Umrah pilgrimage [60, 61]. Such mass gathering events, of millions of Muslims to the holy cities of Makkah and Al-Madinah in western Saudi Arabia, represent unique events which could lead to virus introductions into the kingdom with a high potential for global dissemination of novel reassortants [21,22,23]. Indeed, the prevalence of influenza at Hajj has been suggested to be up to 4.5 times higher than that in the community [22], suggesting that such events could represent a melting pot for co-circulation of influenza viruses from multiple lineages. This could not only reflect the global diversity of these viruses but also result in an increase in reassortment events and the emergence of novel influenza viruses. Thus, vigilant surveillance and monitoring of influenza diversity during these mass gatherings and in surrounding cities such as Jeddah (the second largest city in Saudi Arabia and the most important port of entry into the holy cities of Makkah and Al-Madinah) are of great importance. While the genetic diversity of circulating influenza A/H1N1pdm09 viruses in the current study resembled global genetic diversity, concurrent detection of viruses from clades 6b, 6b.1 and 6b.2 in 2015 suggests multiple introductions into Saudi Arabia from several regions. The observed phylogenetic topologies of all gene segments for these viruses were almost similar with fixed signature amino acid changes, except for the internal genes from clade 6b.2 viruses. Nonetheless, our analysis showed possible intra-subtype reassortment in some viruses such as Saudi Arabia/11/2015, Jeddah/KFAFH0089/2015 and Jeddah/0418/2015, Jeddah/TAM/2015 and Jeddah/9127/2015. Specifically, the NS gene from Saudi Arabia/11/2015 and PB2 genes from Jeddah/KFAFH0089/2015 and Jeddah/0418/2015 isolates were closely related to clade 6b viruses rather than to clade 6b.1 (Figs. 3 and 4). Additionally, the NA and M gene segments from Jeddah/TAM/2015 and Jeddah/9127/2015 fell in clade 6b.1, compared to their other genes which clustered with clade 6b viruses (Figs. 2 and 3).
Continued monitoring and surveillance of influenza viruses in Saudi Arabia, especially in the western region of the country, is very critical as it will help us better understand influenza patterns at both regional and global scales. While the current study has shed some light on the diversity of influenza A/H1N1pdm09 in Saudi Arabia, further and continued studies on a larger number of Saudi isolates are clearly needed, maybe via the establishment of a national influenza surveillance program for the early detection of drug resistant and antigenic variants. This would also contribute to global efforts aimed at the annual selection of influenza vaccines.
References
Medina RA, García-Sastre A (2011) Influenza A viruses: new research developments. Nat Rev Microbiol 9:590–603
Nguyen-Van-Tam JS, Hampson AW (2003) The epidemiology and clinical impact of pandemic influenza. Vaccine 21:1762–1768
World Health Organization (WHO). Influenza (Seasonal) Fact sheet November 2016. http://www.who.int/mediacentre/factsheets/fs211/en/. Accessed 15 Nov 2017
Lui KJ, Kendal AP (1987) Impact of influenza epidemics on mortality in the United States from October 1972 to May 1985. Am J Public Health 77:712–716
Tong S, Zhu X, Li Y et al (2013) New world bats harbor diverse influenza A viruses. PLoS Pathog 9:e1003657
Hay AJ, Gregory V, Douglas AR et al (2001) The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci 356:1861–1870
Martinez O, Tsibane T, Basler CF (2009) Neutralizing anti-influenza virus monoclonal antibodies: therapeutics and tools for discovery. Int Rev Immunol 28:69–92
Schäfer JR, Kawaoka Y, Bean WJ et al (1993) Origin of the pandemic 1957 H2 influenza A virus and the persistence of its possible progenitors in the avian reservoir. Virology 194:781–788
Webby RJ, Webster RG (2001) Emergence of influenza A viruses. Philos Trans R Soc Lond B Biol Sci 356:1817–1828
Garten RJ, Davis CT, Russell CA et al (2009) Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201
Itoh Y, Shinya K, Kiso M et al (2009) In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460:1021–1025
Miller MA, Viboud C, Balinska M et al (2009) The signature features of influenza pandemics—implications for policy. N Engl J Med 360:2595–2598
Smith GJ, Vijaykrishna D, Bahl J et al (2009) Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125
Pan C, Cheung B, Tan S et al (2010) Genomic signature and mutation trend analysis of pandemic (H1N1) 2009 influenza A virus. PLoS One 5:e9549
Cutler J, Schleihauf E, Hatchette TF et al (2009) Investigation of the first cases of human-to-human infection with the new swine-origin influenza A (H1N1) virus in Canada. CMAJ 181:159–163
Fisman DN, Savage R, Gubbay J et al (2009) Older age and a reduced likelihood of 2009 H1N1 virus infection. N Engl J Med 361:2000–2001
Hancock K, Veguilla V, Lu X et al (2009) Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 361:1945–1952
Dorigatti I, Cauchemez S, Ferguson NM (2013) Increased transmissibility explains the third wave of infection by the 2009 H1N1 pandemic virus in England. Proc Natl Acad Sci USA 110:13422–13427
Otte A, Sauter M, Daxer MA et al (2015) Adaptive mutations that occurred during circulation in humans of H1N1 influenza virus in the 2009 pandemic enhance virulence in mice. J Virol 89:7329–7337
Dong G, Peng C, Luo J et al (2015) Adamantane-resistant influenza A viruses in the world (1902–2013): frequency and distribution of M2 gene mutations. PLoS One 10:e0119115
Cobbin JCA, Alfelali M, Barasheed O et al (2017) Multiple sources of genetic diversity of influenza A viruses during the Hajj. J Virol 91:e00096-17
Benkouiten S, Charrel R, Belhouchat K et al (2014) Respiratory viruses and bacteria among pilgrims during the 2013 Hajj. Emerg Infect Dis 20:1821–1827
Memish ZA, Assiri A, Turkestani A et al (2015) Mass gathering and globalization of respiratory pathogens during the 2013 Hajj. Clin Microbiol Infect 21:571.e571–571.e578
Tolah AM, Azhar EI, Hashem AM (2016) Susceptibility of influenza viruses circulating in Western Saudi Arabia to neuraminidase inhibitors. Saudi Med J 37:461–465
World Health Organization (WHO) Sequencing primers WHO protocol. http://www.who.int/csr/resources/publications/swineflu/GenomePrimers_20090512.pdf?ua=1. Accessed 15 Nov 2017
Al-Saeed MS, El-Kafrawy SA, Farraj SA et al (2017) Phylogenetic characterization of circulating Dengue and Alkhumra Hemorrhagic Fever viruses in western Saudi Arabia and lack of evidence of Zika virus in the region: a retrospective study, 2010–2015. J Med Virol 89:1339–1346
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Tamura K, Stecher G, Peterson D et al (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tamura K (1992) Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol Biol Evol 9:678–687
Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16:111–120
Hasegawa M, Kishino H, Yano T (1985) Dating the human-ape split by a molecular clock of mitochondrial DNA. J Mol Evol 22:160–174
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York
Houng HS, Garner J, Zhou Y et al (2012) Emergent 2009 influenza A(H1N1) viruses containing HA D222N mutation associated with severe clinical outcomes in the Americas. J Clin Virol 53:12–15
Nguyen HK, Nguyen PT, Nguyen TC et al (2015) Virological characterization of influenza H1N1pdm09 in Vietnam, 2010–2013. Influenza Other Respir Viruses 9:216–224
Parida M, Dash PK, Kumar JS et al (2016) Emergence of influenza A(H1N1)pdm09 genogroup 6B and drug resistant virus, India, January to May 2015. Euro Surveill 21:6–11
Wedde M, Biere B, Wolff T et al (2015) Evolution of the hemagglutinin expressed by human influenza A(H1N1)pdm09 and A(H3N2) viruses circulating between 2008–2009 and 2013–2014 in Germany. Int J Med Microbiol 305:762–775
European Centre for Disease Prevention and Control. Influenza virus characterisation, summary Europe, November 2015. Stockholm: ECDC; 2015. https://ecdc.europa.eu/sites/portal/files/media/en/publications/Publications/influenza-virus-characterisation-november-2015.pdf. Accessed 15 Nov 2017
Katz J. Global Surveillance and Virus Characterization October 13, 2016. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM525491.pdf. Accessed 15 Nov 2017
Katz J. Global Surveillance and Virus Characterization March 4, 2016. https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/BloodVaccinesandOtherBiologics/VaccinesandRelatedBiologicalProductsAdvisoryCommittee/UCM492316.pdf. Accessed 15 Nov 2017
World Health Organization (WHO). Recommended composition of influenza virus vaccines for use in the 2017 southern hemisphere influenza season. Available from: http://www.who.int/influenza/vaccines/virus/recommendations/201609_recommendation.pdf?ua=1. Accessed 15 Nov 2017
Broberg E, Melidou A, Prosenc K et al (2016) Predominance of influenza A(H1N1)pdm09 virus genetic subclade 6B.1 and influenza B/Victoria lineage viruses at the start of the 2015/16 influenza season in Europe. Euro Surveill 21:pii=30184. https://doi.org/10.2807/1560-7917.ES.2016.21.13.30184
Al-Qahtani AA, Mubin M, Dela Cruz DM et al (2017) Phylogenetic and nucleotide sequence analysis of influenza A (H1N1) HA and NA genes of strains isolated from Saudi Arabia. J Infect Dev Ctries 11:81–88
Yi H, Lee JY, Hong EH et al (2010) Oseltamivir-resistant pandemic (H1N1) 2009 virus, South Korea. Emerg Infect Dis 16:1938–1942
Shin SY, Kang C, Gwack J et al (2011) Drug-resistant pandemic (H1N1) 2009, South Korea. Emerg Infect Dis 17:702–704
Hurt AC, Leang SK, Speers DJ et al (2012) Mutations I117V and I117M and oseltamivir sensitivity of pandemic (H1N1) 2009 viruses. Emerg Infect Dis 18:109–112
Kong W, Liu L, Wang Y et al (2014) Hemagglutinin mutation D222N of the 2009 pandemic H1N1 influenza virus alters receptor specificity without affecting virulence in mice. Virus Res 189:79–86
Matos-Patrón A, Byrd-Leotis L, Steinhauer DA et al (2015) Amino acid substitution D222N from fatal influenza infection affects receptor-binding properties of the influenza A(H1N1)pdm09 virus. Virology 484:15–21
Goka EA, Vallely PJ, Mutton KJ et al (2014) Mutations associated with severity of the pandemic influenza A(H1N1)pdm09 in humans: a systematic review and meta-analysis of epidemiological evidence. Arch Virol 159:3167–3183
Jagger BW, Wise HM, Kash JC et al (2012) An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204
Yewdell JW, Ince WL (2012) Virology. Frameshifting to PA-X influenza. Science 337:164–165
Shi M, Jagger BW, Wise HM et al (2012) Evolutionary conservation of the PA-X open reading frame in segment 3 of influenza A virus. J Virol 86:12411–12413
Kosik I, Holly J, Russ G (2013) PB1-F2 expedition from the whole protein through the domain to aa residue function. Acta Virol 57:138–148
Hayashi T, MacDonald LA, Takimoto T (2015) Influenza A virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J Virol 89:6442–6452
Bavagnoli L, Cucuzza S, Campanini G et al (2015) The novel influenza A virus protein PA-X and its naturally deleted variant show different enzymatic properties in comparison to the viral endonuclease PA. Nucleic Acids Res 43:9405–9417
Gao H, Sun H, Hu J et al (2015) Twenty amino acids at the C-terminus of PA-X are associated with increased influenza A virus replication and pathogenicity. J Gen Virol 96:2036–2049
Xu G, Zhang X, Sun Y et al (2016) Truncation of C-terminal 20 amino acids in PA-X contributes to adaptation of swine influenza virus in pigs. Sci Rep 6:21845
Lee J, Yu H, Li Y et al (2017) Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology 504:25–35
Trifonov V, Racaniello V, Rabadan R (2009) The contribution of the PB1-F2 protein to the fitness of influenza A viruses and its recent evolution in the 2009 influenza A (H1N1) pandemic virus. PLoS Curr 1:RRN1006
General Authority for Statistics, Kingdom of Saudi Arabia. Umrah Survey 2016. https://www.stats.gov.sa/sites/default/files/umrah_surveyen_0.pdf. Accessed 15 Nov 2017
General Authority for Statistics, Kingdom of Saudi Arabia. Hajj Statistics 2016. https://www.stats.gov.sa/sites/default/files/hajj_1437_en.pdf. Accessed 15 Nov 2017
Funding
This study was funded by the Deanship of Scientific Research (DSR), King Abdulaziz University (KAU), Jeddah, under grant no. (G-484-140-37). The authors, therefore, acknowledge with thanks DSR for technical and financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Handling Editor: Hans Dieter Klenk.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hashem, A.M., Azhar, E.I., Shalhoub, S. et al. Genetic characterization and diversity of circulating influenza A/H1N1pdm09 viruses isolated in Jeddah, Saudi Arabia between 2014 and 2015. Arch Virol 163, 1219–1230 (2018). https://doi.org/10.1007/s00705-018-3732-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-3732-y