
Abstract
Infections by H3N2-type influenza A viruses (IAV) resulted in significant numbers of hospitalization in several countries in 2014-2015, causing disease also in vaccinated individuals and, in some cases, fatal outcomes. In this study, sequence analysis of H3N2 viruses isolated in Germany from 1998 to 2015, including eleven H3N2 isolates collected early in 2015, was performed. Compared to the vaccine strain A/Texas/50/2012 (H3N2), the 2015 strains from Germany showed up to 4.5 % sequence diversity in their HA1 protein, indicating substantial genetic drift. The data further suggest that two distinct phylogroups, 3C.2 and 3C.3, with 1.6-2.3 % and 0.3-2.4 % HA1 nucleotide and amino acid sequence diversity, respectively, co-circulated in Germany in the 2014/2015 season. Distinct glycosylation patterns and amino acid substitutions in the hemagglutinin and neuraminidase proteins were identified, possibly contributing to the unusually high number of H3N2 infections in this season and providing important information for developing vaccines that are effective against both genotypes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Annual influenza epidemics are mainly caused by influenza A (subtype H1N1 and H3N2) and influenza B viruses (IAV/IBV). Since their emergence in 1968, which resulted in the third pandemic of the 20th century, H3N2 IAVs have been circulating in the human population. Occasionally, H3N2 strains were even more prevalent than the co-circulating H1N1 IAV and IBV strains [13] and caused high morbidity and mortality rates [2].
The well-established rapid evolution of influenza viruses (IV) is mainly achieved by antigenic shift and antigenic drift [26]. About seven H3N2 clades (designated clades 1 to 7) and many subclades have evolved over the past few years, providing a significant challenge for the annual design of effective influenza vaccines [24]. At present, viruses in clade 3 are the dominant group, which has diversified further into three major subclades designated 3A, 3B, and 3C, with subclade 3C containing several distinguishable genetic groups [9, 24].
The majority of the circulating H3N2 IAVs in late 2014 and early 2015 appear to be genetically and antigenically distinct from influenza virus A/Texas/50/2012 (H3N2, TX12), i.e., the 2014/2015 vaccine strain recommended for the northern hemisphere [4]. In line with this, there is increasing evidence (reported from different countries) for limited protection afforded by the TX12-based vaccine against H3N2 IAVs during the 2014/2015 season, particularly among the elderly [1, 7, 9]. Similar data have also been reported for strains collected in Germany in 2013 [21], but there is limited information on the genetic characteristics and evolution of these German H3N2 IAVs. In the present study, we determined the complete genome sequences of two viruses (H3N22176 and H3N23444) and determined the sequences of the HA, NA and M genes of another nine viruses obtained from hospitalized patients at the university hospital of Giessen, Germany, between January and April 2015. Our study provides insight into specific genetic markers, phylogenetic relationships and evolution of these H3N2 viruses from Giessen compared to other H3N2 IAV isolates collected in Germany, other European countries, and other continents.
Materials and methods
Sample preparation and typing
Nasopharyngeal swabs were collected from patients with respiratory infections and further processed by the Virus Diagnostics Laboratory at the Institute of Medical Virology (Justus Liebig University Giessen, Germany). Swabs were washed with 800 µl of 0.9 % NaCl solution. Three hundred microliters of each sample was subjected to analysis using a Gene Xpert IV system (Cepheid, USA), using the Xpert Flu/RSV XC test according to the manufacturer’s instructions. In parallel, viral RNA was extracted from 400 µl of each sample using an EZ1 Advanced XL system (QIAGEN, Germany). Ten microliters of the resulting 60-µl eluate fraction was used in the xTAG Respiratory Viral Panel Fast v2 (Luminex 200 platform; Luminex, The Netherlands) to determine influenza virus types/subtypes as well as possible coinfections with other respiratory viruses. The data collection and publication were performed in accordance with the data protection law of the state of Hesse (HDSG, §33). All experiments were performed in a biosafety level 2 (BSL2) containment laboratory approved for such use by the local authorities (RP, Giessen, Germany).
Viral genome sequencing and sequence data collection
Full-gene amplification of the viral segments was done using appropriate oligonucleotide primers as described previously [10]. Amplicons were cloned into the vector pMPccdB as described previously [17] and sequenced. The sequences determined for the HA, NA and M segments of nine isolates and full genome sequences of two isolates were submitted to the Global Initiative on Sharing All Influenza Data (GISAID) (Table 1). Sequences of viruses isolated in Germany from 1998 to April 2015 and viruses from Africa, Asia, Europe, South America and North America were retrieved from the GenBank and GISAID databases and used for the construction of phylogenetic trees.
Phylogenetic analysis and evolution rate
Nucleotide and deduced amino acid (aa) sequences were aligned using MAFFT [11] and further edited using BioEdit [8]. A total of 813 gene sequences were processed for genetic analysis (Table 2). Numbering of aa residues of the HA (H3 numbering after removal of the signal peptide) and other proteins (open reading frame) was based on that of the TX12 strain. The HA and NA sequences were also compared with those of influenza A/Switzerland/9715293/2013 (designated hereafter as CH13), which was recently recommended for use as vaccine for the 2015/2016 season [1].
Phylogenetic analysis was performed using Topali v2.5 [16] after selection of the best-fit model of nucleotide substitution. Estimated origin dates of H3N2 viruses were determined using the Bayesian Markov chain Monte Carlo (MCMC) method implemented in BEAST v1.6.1 [3]. The evolution rate and time from the most recent common ancestor (tMRCA) were estimated using a Bayesian Skyline coalescent tree [3] prior to selecting a relaxed uncorrelated lognormal model with gamma-distributed rate heterogeneity. tMRCA was estimated only for the HA1 and NA genes using 145 sequences of human origin with the collection date adjusted to year-month-day format. MCMC chains were run for 5-10 × 107 generations for each dataset, with 10 % burn-in and 95 % highest probability density (HPD) values. TRACER v1.6 was used for the analysis of MCMC output [3]. TreeAnnotator v1.8 (http://beast.bio.ed.ac.uk/TreeAnnotator) and FigTree v1.4 (http://beast.bio.ed.ac.uk/figtree) were used to generate and visualize trees, which were further edited using Inkscape.
Prediction of glycosylation
Potential asparagine-linked glycosylation sites (PNGs) in the HA and NA were predicted using the NetNGlyc 1.0 Server to examine the sequence context of N–X–S/T motifs, where X can be any amino acid except proline (http://www.cbs.dtu.dk/services/NetNGlyc/).
Estimation of positive selection
To assess whether Giessen isolates underwent episodic/positive diversifying selection, the calculated non-synonymous/synonymous (dN/dS) ratio at each codon in the alignment was assessed using Datamonkey, a webserver of the HyPhy package, using the best-fit substitution model as recommended [6, 20]. Four different codon-based maximum-likelihood methods at posterior probabilities ‘pp’ of 0.05 were used: the mixed-effects model of evolution (MEME), the most conservative “single-likelihood ancestor counting” algorithm (SLAC), “fixed effects likelihood” (FEL) and internal FEL (IFEL) [20].
3D structural modeling
Three-dimensional structures (3D) of the HA and NA monomers of the Giessen isolates were compared with those obtained for the TX12 strain. Distinct mutations in the HA and NA of the Giessen isolates were superimposed on the predicted 3D structure of TX12 using Geneious version 8.1.5 [12] and edited further manually.
Results
Epidemiology
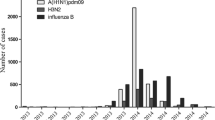
From December 2014 to May 2015, 85 patients were treated for (laboratory-confirmed) influenza in the Giessen and Marburg University Hospital (Campus Giessen), Hesse, Germany. Among these patients, 75 (88 %; 41 males and 34 females) tested positive for IAV, and 10 (12 %; 6 males and 4 females) tested positive for IBV. To further characterize these viruses, a total of 38 (out of 75) IAV isolates were subjected to subtype analysis. In 27 cases (14 male and 13 female patients; median age = 42.6) subtype H3N2 was identified, while in 11 cases (8 male and 3 female patients; median age = 51.3), the 2009 pandemic H1N1 (H1N1pdm2009) IAV was identified. Infections with H1N1pdm2009 IAVs were significantly more frequent in elderly patients (aged 51-70 years), while H3N2 infections were detected with similar frequencies in all age groups. In the time period between December 2014 and March 2015, H3N2 infections were significantly more frequent than H1N1pdm2009 and IBV infections, with a peak of H3N2 infections occurring in February 2015 (data not shown). To further characterize the H3N2 IAVs involved in these infections, we selected 11 H3N2 isolates (five isolates from females [aged 12-78 years] and six from males [aged 23-86 years]) that we collected between February and March 2015 (Table 1).
Sequence analysis
HA and NA gene sequences determined in this study (Table 1) were compared to those of the vaccine strains TX12 and CH13 (influenza A/Switzerland/9715293/2013, which is to be used for the 2015/2016 vaccine [1]). In the 11 protein sequences determined for the Giessen isolates, a total of 16 amino acid sequence variations were identified (Table 2).
Genome segments that encode viral surface proteins
Hemagglutinin (H3 HA)
The HA of the 11 viruses analyzed in this study shared 97.4-99.7 % and 97.4-100 % nucleotide and aa sequence identity, respectively. Compared to TX12, they had 98.1-98.4 % and 98-99.1 % nucleotide and aa sequence identity, respectively. The HA1 domain of the 2015 Giessen strains analyzed in this study (n = 63) had up to 2.3 % nucleotide and 5.7 % aa sequence diversity, whereas HA1 of the German strains from 2014 (n = 35) had up to 2 % nucleotide and 4.7 % aa sequence diversity. The HA1 of the 2015 Giessen isolates showed 97.7 % nucleotide and 95.5 % aa sequence identity compared to TX12, while the HA1 of the 2014 isolates showed 98 % nucleotide and 96.2 % aa sequence identity. Compared to TX12, all Giessen strains (as well as CH13) contained three unique replacements in their HA proteins: N128A/T, V186G, and F219S. Furthermore, all strains except H3N23232 were found to carry a P198S substitution. In addition, 71 % of the 2015 isolates (including 9 out of the 11 Giessen isolates characterized in this study) and <50 % of the 2013/2014 isolates had another six specific replacements (L3I, N144S, F159Y, N225D, Q311H and D489N). With just one exception (see below), the latter six substitutions were not observed in any of the German H3N2 isolates collected in 2012 (n = 24) and 2013 (n = 6) (data not shown). In one (out of 24) of the 2012 isolates, the L3I and D489N replacements were identified in the HA sequence. Compared to CH13, all 2014/2015 viruses but one from 2014 gained S138A, S159F/Y and R326K.
Neuraminidase (NA)
The N2-NA sequences of the isolated strains revealed sequence identities of 98.0-98.4 % at nucleotide level and 98.0-99.1 % at aa level compared with TX12. Furthermore, except for one isolate, all NA sequences from German viruses collected between 2013 and 2015 (including the Giessen isolates of the present study), contained an H150R replacement compared to TX12. Moreover, E221D was observed only in viruses from 2015 (84 %; n = 27/32) and 2014 (62 %; n = 21/34). Likewise, T267K and I380V were detected in 21 (66 %) and 20 (63 %) of the 2015 H3N2-IAVs, respectively, whereas, both T267K and I380V were less abundant and evenly distributed (26 %) in the NA of 2014 H3N2-IAVs (n = 9/34). Interestingly, none of H3N2 IAV isolates collected in 2012 and 2013 carry these changes. Compared to CH13, all 2015 viruses and only five of the 2014 viruses display T392I/M.
Other gene segments
Other segments shared 97.7-99.6 % nucleotide and 95.5-100 % aa sequence identity with TX12 and 98.4-100 % aa sequence identity with each other. The PB2 proteins of H3N22176 and H3N23444 differed from TX12 by two aa replacements (V63I and I588T). With regard to the PB1 protein, H3N22176 was 100 % identical to the vaccine strain, while H3N23444 carried two aa replacements (M111K and F251L). The PB1-F2 of H3N23444 (90 aa) carried a unique I18T substitution. Concerning PA, both H3N22176 and H3N23444 differed from TX12 at three positions (N274S, V670I, and N677K). The NP of H3N22176 and H3N23444 carried an R384G replacement, while H3N22176 had an M331I substitution. The M1 protein was highly conserved among the Giessen isolates. Seven out of 11 were found to be 100 % identical to TX12, while the other four strains had one or two aa substitutions, resulting in aa sequence identities of ≥99.2 %. Moreover, three of these four strains (H3N22390, H3N23448 and H3N22276) had an S120A substitution in their M1 protein. The M2 protein of all 11 Giessen isolates and TX12 contained the amantadine resistance marker S31N. The NS1 proteins of H3N22176 and H3N23444 (230 aa) had a human-IAV like PDZ domain (226RSEV230) and two unique replacements (E26K and Q193R). NEP of H3N22176 and H3N23444 were found to share a K35E replacement compared to TX12 (Table 2).
Variation in the potential N-glycosylation sites (PNGs)
With respect to HA, 10-11 PNGs were predicted to be present in the 11 Giessen isolates, while CH13 has 12 PNGs. Nine PNGs (8NST10, 22NGT24, 38NAT40, 45NSS47, 133NGT135, 165NVT167, 246NST248, 285NGS287, 483NGT485) were conserved in all isolates. Furthermore, all isolates except H3N23940 contained 63NCT65, and all except H3N23448 and H3N22276 contained the 122NES124 site. The latter two viruses as well as CH13 also contained 144NNS146. Regarding NA, 7-9 PNGs were predicted for the Giessen isolates. Of these, five PNGs were conserved across all isolates (70NTT72, 146NNT148, 200NAT202, 234NGT236, 367NET369), while the remaining two sites, 61NIT63 and 86NWS88, were conserved in all but one (H3N22108) isolate. Furthermore, except for H3N24015, the 329NDS331 site was strictly conserved, while only three isolates (H3N23444, H3N22749 and H3N22108) contained the 245NAS247 site.
Phylogenetic analysis
Hemagglutinin
The H3-HA of the Giessen isolates and a selection of viruses from Europe, Asia, Africa and the Americas (the latter isolated in 2014/2015) could be assigned to two distinct groups in clade 3C, called 3C.2 and 3C.3 (Fig. 1), that likely emerged in the first trimester of 2011 as indicated by tMRCA. Group 3C.3 contains the majority of the 2015 German isolates (70 %; n = 44/63) and 43 % of the available 2014 viruses (n = 15), while group 3C.2 contains 30 % of the 2015 German isolates (n = 19/63) and the majority of the 2014 isolates (57 %; n = 20/35). Nine out of 11 sequences determined in this study were assigned to 3C.2, while only H3N22276 and H3N23448 belonged to group 3C.3. Both groups are distinct from TX12, which belongs to group 3C.1 (Fig. 1). Moreover, clade 3C.3 (including the CH13 strain) is distinct from TX12, as shown in Fig. 1. Importantly, clade 3C.3 includes two genetically distinct subgroups (designated 3C.3a and 3C.3b), which are distinguishable from TX12 and also differ substantially from CH13. The H3N2 IAVs of clade 3C.3 differ from TX12 by 6 aa (N128A, R142G, N145S, V186G, P198S and F219S). Unlike TX12 and CH13, the H3N2 IAVs in 3C.3a possess three unique aa substitutions (L157S, I214, and V347K), those in 3C.3b have six unique aa substitutions (E62K, K83R, N122D, L157S, R261Q, V347K), and those in subclade 3C.3b2 harbor an additional unique substitution, Q197H. The 3C.3b viruses also differed from CH13 by S138A, S159F, D225N, and R326K (Fig. 1).

Phylogenetic relationship between the HA genes of H3N2 viruses and vaccine strains. The H3 HA sequences of recent H3N2 IAVs collected in Europe (including the Giessen isolates), Asia, Africa, and the Americas from 2014/2015 can be assigned to two distinct groups: 3C.2 and 3C.3. The majority of the viruses analyzed in this study (red) belong to group 3C.2 (depicted in red), while (only) two viruses (2276 and 3448) belong to group 3C.3 (depicted in blue). Both groups are clearly distinct from the vaccine strains TX12 (subclade 3C.1) and CH13 (subclade 3C.3). Subgroups are indicated at the right. Amino acid substitutions present in specific subgroups and distinct from TX12 are shown in standard font, while substitutions that differ from both vaccine strains are shown in boldface. Maximum-likelihood trees were generated using Topali v2.5 with 1000 bootstrap replicates and further edited using FigTree and Inkscape
Regarding subclade 3C.2, H3N2 IAVs within this clade are distinct from TX12 by 12 aa (L3I, A/N128T, N144S, S/F159Y, K160T, Q311H, D489N N145S, V186G, P198S, F219S and N225D) and from CH13 by 10 aa (L3I, A/N128T, N144S, S/F159Y, K160T, Q311H, D489N, S138A, G142R and R326K).
Neuraminidase
The NA phylogeny differed from that of the HA (Fig. 2). The selected NA sequences of 2014/2015 H3N2 IAVs in Europe, Asia, Africa and the Americas were assigned to two distinct groups (designated here as A and B) that, again, are thought to have emerged in the first trimester of 2011, as indicated by tMRCA. Group A is a 3B HA-like group, while group B corresponds to the 3C group of the HA. Group A comprises only four Giessen isolates from 2015, 12 viruses from 2014, and a few isolates from 2011-2013, whereas group B contains the majority of viruses from 2011 to 2015 as well as the vaccine strains. Compared to both vaccine strains, group A possesses Y155F, D251V and S315G, with two additional variations, D221E and T392I, from CH13. Group B, which corresponds to the HA 3C group (see above), contains TX12 and two distinct B2 and B3 subgroups that probably co-evolved in early 2013 and are most similar to the HA 3C.2 and 3C.3 group, respectively. Four Giessen isolates (H3N22390, H3N22276, H3N23448 and H3N23232) fall into the small B2 subgroup characterized by M51V, V143G, V263I, and T434N substitutions, which separate them from both vaccine strains, and an additional aa variation (T392I) compared to CH13. The majority of the Giessen isolates (7/11) fell in subgroup B3 (subgroups B3.1 and B3.2). The smallest group, B3.1, containing five 2014 viruses as well as CH13, has the unique I392T replacement compared to TX12. Viruses in the B3.2 group, which contains the majority of the 2014-2015 German isolates, differ from TX12 and CH13 by the aa substitutions T267K and I380V (except H3N22176) and by an additional aa variation (T392I) from CH13.

Phylogenetic relationship between the NA genes of H3N2 viruses and vaccine strains. The N2 NA sequences of recent H3N2 IAVs collected in Europe (including the Giessen isolates), Asia, Africa, and the Americas from 2014/2015 can be assigned to two distinct groups (A and B). The majority of viruses analyzed in this study belong to group B3 (depicted in red), while only two viruses (2276 and 3448) were found to belong to group B2 (shown in blue). Both groups are clearly distinct from the vaccine strains TX12 and CH13
Matrix protein
The M gene phylogeny was similar to the phylogenies determined for “internal” viral proteins (see below). Subgroup 1.1 contains eight out of the 11 strains characterized in this study along with TX12. Subgroup 1.2, although not supported by high bootstrap values, contains H3N22390, H3N22276 and H3N23448. Group 1 also contains a third subgroup, 1.3, which includes German viruses isolated between 2004 and 2006. Group 2 consists mainly of German isolates collected between 1998 and 2006 (data not shown).
Other gene segments
Analysis of the PB2, PB1, PA, NP and NS genes revealed two distinct phylogenetic groups (data not shown). Group 1 contained viruses circulating from the late 1990s until 2004 while group 2 mainly contained viruses from Germany (most of which were isolated more recently) that can be further subdivided into two subgroups (2.1 and 2.2). Subgroup 2.1 contains the two viruses sequenced in this study, which cluster with other contemporary viruses from different countries as well as TX12. Group 2.2 contains recent viruses from Europe, America, and Asia.
Positive selection and evolution rates
As shown in Table 2, PB1-F2 was the only gene that was shown to be under positive selection, which, however, may be due to the small sample size for this protein (n = 16). The highest numbers of aa residues under positive selection were detected in the NA (12 sites), HA1 (7 sites), M1 (7 sites) and PB1-F2 (5 sites) proteins. The evolution rate of the German H3N2 isolates was estimated to be 7.5 × 10−3 substitution/site/year for the HA1 and 4.5 × 10−3 substitution/site/year for the NA gene.
Tertiary structure
In the HA of all Giessen isolates, four unique mutations (N128A/T, N145S, V186G, and F219S) where identified. All of these residues are part of an exposed surface of the HA1 head domain (Fig. 3) surrounding the receptor-binding pocket. In addition, the HA proteins of 9 out of the 11 viruses analyzed in this study contain the aa substitutions L3I, N144S, F159Y, N225D, Q311H, and D489N. N/S144, F/Y159, N/D225 are part of the head domain, while the Q/H311 residue is located in the HA1 stalk region. D489N is the only replacement in HA2, where it is located close to the C-terminal end of the protein. In the NA, V143G, H150R, E221D, V263I, T267K and I380V are on the surface of the head domain, while M51V is part of the stalk domain. H150R is located close to the active site of the sialidase (Fig. 3).

Predicted tertiary structure of the H3 HA and N2 NA. a Predicted tertiary structure of the HA monomer. Most aa substitutions affect the globular head region and are in close proximity to the receptor-binding pocket. Only Q311H and D489N are located in the stalk domain. b Front (left) and back (right) views of the NA head domain of H3N2 with positions of aa substitutions in the Giessen isolates indicated
Discussion
Influenza A/H3N2-type viruses were first recognized as major human pathogens in 1968 and continue to co-circulate with other H1N1 IAVs and IBVs in the human population [2]. The incidence of H3N2-associated infections reached its peak early in 2015 and also caused disease in vaccinated individuals [4]. In this study, we analyzed 43 genome segments from eleven H3N2 isolates to obtain insight into the evolution and of German H3N2 viruses collected in early 2015 their phylogenetic relationship to other H3N2 IAVs circulating in Europe, Asia, Africa and the Americas.
The HA of the H3N2 viruses from 2014/2015 isolated in Giessen/Germany clustered with the recent strains from Europe, Asia, Africa and Americas in two distinct phylogroups that are clearly distinguishable from TX12 and, to a lesser extent, from CH13. The phylogenetic topology of HA did not correlate with that of the NA gene segment in these isolates, and the evolutionary rate of HA1 was found to be much higher than that of the NA, suggesting that (multiple) genetic reassortment events between viruses from the 3C.3b and 3B and/or other subgroups played a major role in the emergence of those recently emerging H3N2 IAVs, while co-evolution of HA and NA pairs was probably less important for retaining viral fitness in this case. The observed discordance between the phylogeny and mutational patterns of the HA and NA proteins in the German H3N2 IAVs is consistent with a study by Sandbulte et al. [22] reporting on discordant antigenic shifts of NA and HA in H1N1 and H3N2 IAVs.
Our data provide evidence for a significant genetic drift being involved in the emergence of the recent German H3N2 IAVs and their increasing phylogenetic distance from the vaccine strain TX12. Several replacements in the HA and NA proteins appeared only very recently, starting in 2014 and became more predominant in 2015. Our study identifies a number of aa substitutions in the antigenic sites A to E of the Giessen H3N2 IAVs that may help the virus to escape from antibodies specific for TX12 and/or CH13 [5] (Table 3). Also, aa substitutions in or adjacent to the receptor binding domain (RBD), especially at position 225 and, to a lesser extent, at positions 186, 187, 198, 214 and 219, may modulate the affinity of the 2015 H3N2-IAVs isolates from Germany to host-cell receptors [14].
Glycosylation and deglycosylation is an important viral mechanism to (i) mask antigenic epitopes and thus prevent neutralization by the humoral immune response [15], (ii) adjust receptor-binding affinity [19], or (iii) modulate virulence of IAV in mammals [23]. Although the German H3N2 isolates shared at least nine PNGs with the vaccine strains TX12 and CH13, their glycosylation patterns were not identical. For example, H3N2 IAVs in group 3C.3b had lost the glycosylation site 122NES124 (present in CH13 and the 3C.3 viruses) and acquired another site at 144NNS146, whereas (most of) the 3C.3 viruses had the 63NCT65 site. Therefore, the HA glycosylation patterns predicted may also contribute to a potential antigenic escape of H3N2 IAVs from antibodies raised against the currently used vaccine strains and/or increase viral replication by yet-to-be-characterized mechanisms [5].
There is a meticulously controlled functional balance between viral entry (mediated by HA) and efficient release of virus progeny from the cell surface, with NA playing a major role. Therefore, mutations in the HA are often associated with specific “compensatory” mutations in the NA [28]. Possible functional implications of the substitutions identified in NA in the 2015 Giessen H3N2 isolates remain to be determined. However, it should be noted that residue 150 is located in the 150-loop, which is part of the catalytic site of the enzyme and has been shown previously to affect the enzymatic activity of the NA of H3N2 viruses [27]. Likewise, 221 and 155 are very close to the active site of the enzyme [18]. It remains to be seen if some of these substitutions also affect the immunogenic properties of the mutated NA, even though this may have less-profound effects on protein function than in HA. Giessen H3N2 IAVs have aa substitutions in the dominant NA epitope B (position 221) [18] and epitope A (position 380) [18], both of which may be involved in virus escape from anti-NA antibodies.
We also detected unusual aa substitutions in several internal proteins of the Giessen H3N2 strains. To date, the potential functional consequences of these substitutions are not known in most cases. Interestingly, R384K in the NP is part of a CTL epitope, and mutations at this site have been reported to abrogate recognition of a H3N2 virus by cytotoxic T-lymphocytes [25], suggesting a possible role of this particular substitution in virus escape from the cellular immune response.
Taken together, this study leads us to suggest that specific aa substitutions in the 2015 Giessen H3N2 IAV isolates, particularly in the immunogenic determinants and glycosylation sites of HA and NA may have contributed to inefficient protection after vaccination with the TX12 strain. Some of these genetic changes may also impair the efficacy of the CH13-based vaccine for raising a protective immune response against a diverse group of H3N2 IAV strains circulating in Germany. In this context, the development of modified CH13-like vaccines based on the 3C.3 clade or bivalent 3C.2/3C.3-based vaccines may be considered.
References
Appiah GD, Blanton L, D’Mello T, Kniss K, Smith S, Mustaquim D, Steffens C, Dhara R, Cohen J, Chaves SS, Bresee J, Wallis T, Xu X, Abd Elal AI, Gubareva L, Wentworth DE, Katz J, Jernigan D, Brammer L (2015) Influenza activity—United States, 2014–15 season and composition of the 2015–16 influenza vaccine. MMWR Morb Mortal Wkly Rep 64:583–590
Bedford T, Riley S, Barr IG, Broor S, Chadha M, Cox NJ, Daniels RS, Gunasekaran CP, Hurt AC, Kelso A, Klimov A, Lewis NS, Li X, McCauley JW, Odagiri T, Potdar V, Rambaut A, Shu Y, Skepner E, Smith DJ, Suchard MA, Tashiro M, Wang D, Xu X, Lemey P, Russell CA (2015) Global circulation patterns of seasonal influenza viruses vary with antigenic drift. Nature 523:217–220
Bouckaert R, Heled J, Kuhnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ (2014) BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537
Broberg E, Snacken R, Adlhoch C, Beauté J, Galinska M, Pereyaslov D, Brown C, Penttinen P; WHO European Region and the European Influenza Surveillance Network (2015) Start of the 2014/15 influenza season in Europe: drifted influenza A (H3N2) viruses circulate as dominant subtype. Euro Surveill 20(4)
Chambers BS, Parkhouse K, Ross TM, Alby K, Hensley SE (2015) Identification of hemagglutinin residues responsible for H3N2 antigenic drift during the 2014–2015 influenza season. Cell Rep 12:1–6
Delport W, Poon AF, Frost SD, Kosakovsky Pond SL (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457
Gilca R, Skowronski DM, Douville-Fradet M, Amini R, Boulianne N, Rouleau I, Martineau C, Charest H, De Serres G (2015) Mid-season estimates of influenza vaccine effectiveness against influenza A (H3N2) hospitalization in the elderly in Quebec, Canada, January 2015. PloS One 10:e0132195
Hall T (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Haveri A, Ikonen N, Julkunen I, Kantele A, Anttila V, Ruotsalainen E, Nohynek H, Lyytikainen O, Savolainen-Kopra C (2015) Reduced cross-protection against influenza A(H3N2) subgroup 3C.2a and 3C.3a viruses among Finnish healthcare workers vaccinated with 2013/14 seasonal influenza vaccine. Euro Surveill: bulletin Europeen sur les maladies transmissibles Eur Commun Dis Bull 20:21028
Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR (2001) Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol 146:2275–2289
Katoh K, Standley DM (2014) MAFFT: iterative refinement and additional methods. Methods Mol Biol 1079:131–146
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Lin J, Kang M, Zhong H, Zhang X, Yang F, Ni H, Huang P, Hong T, Ke C, He J (2013) Influenza seasonality and predominant subtypes of influenza virus in Guangdong, China, 2004–2012. J Thorac Dis 5(Suppl 2):S109–S117
Lin YP, Xiong X, Wharton SA, Martin SR, Coombs PJ, Vachieri SG, Christodoulou E, Walker PA, Liu J, Skehel JJ, Gamblin SJ, Hay AJ, Daniels RS, McCauley JW (2012) Evolution of the receptor binding properties of the influenza A (H3N2) hemagglutinin. Proc Natl Acad Sci USA 109:21474–21479
Long J, Bushnell RV, Tobin JK, Pan K, Deem MW, Nara PL, Tobin GJ (2011) Evolution of H3N2 influenza virus in a guinea pig model. PloS One 6:e20130
Milne I, Lindner D, Bayer M, Husmeier D, McGuire G, Marshall DF, Wright F (2009) TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics 25:126–127
Mostafa A, Kanrai P, Ziebuhr J, Pleschka S (2013) Improved dual promotor-driven reverse genetics system for influenza viruses. J Virol Methods 193:603–610
Munoz ET, Deem MW (2005) Epitope analysis for influenza vaccine design. Vaccine 23:1144–1148
Owen RE, Yamada E, Thompson CI, Phillipson LJ, Thompson C, Taylor E, Zambon M, Osborn HM, Barclay WS, Borrow P (2007) Alterations in receptor binding properties of recent human influenza H3N2 viruses are associated with reduced natural killer cell lysis of infected cells. J Virol 81:11170–11178
Pond SL, Frost SD, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679
Remschmidt C, Rieck T, Bodeker B, Wichmann O (2015) Application of the screening method to monitor influenza vaccine effectiveness among the elderly in Germany. BMC Infect Dis 15:137
Sandbulte MR, Westgeest KB, Gao J, Xu X, Klimov AI, Russell CA, Burke DF, Smith DJ, Fouchier RA, Eichelberger MC (2011) Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc Natl Acad Sci USA 108:20748–20753
Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL (2002) Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol 40:3256–3260
Stucker KM, Schobel SA, Olsen RJ, Hodges HL, Lin X, Halpin RA, Fedorova N, Stockwell TB, Tovchigrechko A, Das SR, Wentworth DE, Musser JM (2015) Haemagglutinin mutations and glycosylation changes shaped the 2012/13 influenza A(H3N2) epidemic, Houston, Texas. Euro Surveill 20(18)
Voeten JT, Bestebroer TM, Nieuwkoop NJ, Fouchier RA, Osterhaus AD, Rimmelzwaan GF (2000) Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J Virol 74:6800–6807
Wong SS, Webby RJ (2013) Traditional and new influenza vaccines. Clin Microbiol Rev 26:476–492
Wu Y, Qin G, Gao F, Liu Y, Vavricka CJ, Qi J, Jiang H, Yu K, Gao GF (2013) Induced opening of influenza virus neuraminidase N2 150-loop suggests an important role in inhibitor binding. Sci Rep 3:1551
Yen HL, Liang CH, Wu CY, Forrest HL, Ferguson A, Choy KT, Jones J, Wong DD, Cheung PP, Hsu CH, Li OT, Yuen KM, Chan RW, Poon LL, Chan MC, Nicholls JM, Krauss S, Wong CH, Guan Y, Webster RG, Webby RJ, Peiris M (2011) Hemagglutinin-neuraminidase balance confers respiratory-droplet transmissibility of the pandemic H1N1 influenza virus in ferrets. Proc Natl Acad Sci USA 108:14264–14269
Acknowledgments
We acknowledge the authors and originating and submitting laboratories of the sequences from GISAID’s EpiFlu Database on which this research is based. All submitters of data may be contacted directly via the GISAID website http://www.gisaid.org. This work was supported in part by the BMBF-founded FluResearchNet (Molecular Signatures Determining Pathogenicity and Species Transmission of Influenza A Viruses [01 KI 07136 to S.P.]), a fellowship from the German-Egyptian Research Long-Term Scholarship “GERLS” program co-funded by the Egyptian government and the German Academic Exchange Service (DAAD, to A. M.), and the DFG-funded SFB/TR 84 (Innate Immunity of the Lung: Mechanisms of Pathogen Attack and Host Defence in Pneumonia, TP B2 to S. P.) and SFB 1021 (RNA viruses: RNA metabolism, host response and pathogenesis, TP C1 and TP A1 to S.P. and J.Z.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors
Additional information
A. Mostafa and E.-S. M. Abdelwhab equally contributed as first author.
Rights and permissions
About this article

Cite this article
Mostafa, A., Abdelwhab, ES.M., Slanina, H. et al. Phylogenetic analysis of human influenza A/H3N2 viruses isolated in 2015 in Germany indicates significant genetic divergence from vaccine strains. Arch Virol 161, 1505–1515 (2016). https://doi.org/10.1007/s00705-016-2815-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-016-2815-x