
Abstract
We report the first genome sequence of a Colocasia bobone disease-associated virus (CBDaV) derived from bobone-affected taro [Colocasia esculenta L. Schott] from Solomon Islands. The negative-strand RNA genome is 12,193 nt long, with six major open reading frames (ORFs) with the arrangement 3′-N-P-P3-M-G-L-5′. Typical of all rhabdoviruses, the 3′ leader and 5′ trailer sequences show complementarity to each other. Phylogenetic analysis indicated that CBDaV is a member of the genus Cytorhabdovirus, supporting previous reports of virus particles within the cytoplasm of bobone-infected taro cells. The availability of the CBDaV genome sequence now makes it possible to assess the role of this virus in bobone, and possibly alomae disease of taro and confirm that this sequence is that of Colocasia bobone disease virus (CBDV).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
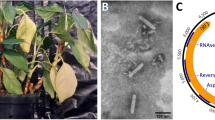
Bobone and alomae are diseases of taro [Colocasia esculenta (L.) Schott] that are restricted to Solomon Islands and Papua New Guinea and whose etiology remains unclear [1, 4, 8, 10]. Two types of rhabdovirus-like particles were observed in infected taro plants; the smaller (~210 × 65 nm) were associated with the characterised taro vein chlorosis virus (TaVCV), while the larger (~300-350 × 50-55 nm), which were associated with severe stunting and gall formation typical of bobone/alomae disease, were those of Colocasia bobone disease virus (CBDV) [8]. We report for the first time the genome sequence and structure of a rhabdovirus isolated from bobone-infected taro plants from Solomon Islands that had the larger virus particles, as well as its phylogenetic relationships to other plant rhabdoviruses. As we have not yet been able to establish that this virus causes bobone disease, we have named this virus Colocasia bobone disease-associated virus (CBDaV).
Taro plants with symptoms of bobone disease were obtained in 1995 from Solomon Islands and continuously propagated in a glasshouse at the University of Auckland, New Zealand, since that date. These plants were subsequently found to also be infected with the potyvirus dasheen mosaic virus (DsMV). Total RNA was isolated from leaves of three taro plants using a SpectrumTM Plant Total RNA Kit, including on-column DNase treatment (Sigma Aldrich). Transcriptome sequencing was carried out for each plant by Macrogen Inc. (South Korea) using HiSeq2000 with transcriptome assembly by New Zealand Genomics Limited. In order to remove potyviral sequences, the taro sequences were pooled and aligned against the DsMV genome (NC_003537) [6]. Reads that did not map to DsMV were extracted using Samtools [7] and assembled de novo using Trinity [2]. To remove any remaining pestiviral sequence, the assembled contigs were BLAST searched against the genomes of DsMV, soybean mosaic virus (FJ640956), turnip mosaic virus (NC_002509), and watermelon mosaic virus (HQ384216). The remaining contigs were queried against the GenBank database using the BLASTn algorithm.
Two contigs of 2,663 and 9,581 bp, which overlapped by 43 bp, were found to be similar to the 3′ and 5′ regions, respectively, of northern cereal mosaic virus (NCMV). The sequence of this overlapping region was confirmed by reverse transcription polymerase chain reaction (RT-PCR) using the primers CBDVF1 (5′-GAGCCAAACTGTCAAAAGC-3′) and CBDVR2 (5′-CGATGCACTGGTCTTGATC-3′) and dideoxy sequencing of the PCR product, which was carried out by the Waikato University Sequencing Facility. The ends of the CBDaV genome were obtained using the 3′ and 5′ RACE System for Rapid Amplification of cDNA ends (Life Technologies). The genome structure of CBDaV was determined by comparison with the following cytorhabdovirus and nucleorhabdovirus genomes: lettuce necrotic yellows virus (LNYV, NC_007642), barley yellow striate mosaic virus (BYSMV, KM213865), NCMV (AB030277 and GU985153), alfalfa dwarf virus (ADV, KP205452), lettuce yellow mottle virus (LYMoV, NC_011532), persimmon virus A (PeVA, NC_018381), TaVCV, (NC_006942), eggplant mottled dwarf virus (EMDV, KJ082087), potato yellow dwarf virus (PYDV, GU734660), maize mosaic virus (MMV, AY618418), maize Iranian mosaic virus (MIMV, DQ186554), maize fine streak virus (MFSV, AY618417), rice yellow stunt virus (RYSV, AB011257), and Sonchus yellow net virus (SYNV, L32603). Nucleotide and amino acid sequences were aligned using Muscle in GeneiousPro v6.1.8 (http://www.geneious.com, [5]), and phylogenetic analysis by maximum likelihood was carried out using MEGA 6.0 [11]. Statistical support for tree branching patterns was determined by bootstrapping one thousand times. Individual protein properties were predicted using the Expasy Compute pI/MW tool and searching against the pfam database.
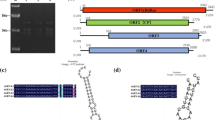
The complete CBDaV genomic RNA (accession number KT381973) is 12,193 nt long with six major ORFs identified in the anti-genomic strand. Based on sequence similarity to proteins encoded by other rhabdoviruses, the CBDaV genome organisation is 3′ leader-N-P-P3-M-G-L-5′ trailer (Supplementary Tables 1 and 2), where N is the nucleocapsid gene, P the phosphoprotein gene, P3 the putative movement protein gene, M the matrix protein gene, G the glycoprotein gene, and L the polymerase gene. This genome arrangement is identical to that of LNYV, the type member of the genus Cytorhabdovirus; however, the most similar sequences based on BLASTX analyses were either NCMV (N and P proteins), or BYSMV (P3, M, G, and L proteins). The L polymerase was the only protein to have any conserved domains, namely pfam00946 (Mononegavirales RNA-dependent RNA polymerase) between amino acids 197 and 1045 (7.59 e-151) and pfam14318 (Mononegavirales mRNA-capping region) between amino acids 1151-2035 (5.65 e-61). Predicted molecular weights and pI values for each CBDaV protein are provided in Supplementary Table 3.
As is typical for all rhabdoviruses, the coding region of the CBDaV genome is flanked by untranslated 3′ leader and 5′ trailer regions that are 177 and 260 nt long, respectively, and which are complementary to each other (data not shown) [3, 9, 12–14]. Like BYSMV, but unlike other cytorhabdoviruses [3, 15], the 3′ leader sequence of CBDaV does not possess a 3′ overhang of 1-2 nucleotides. Analysis of the untranslated sequences highlighted a conserved region similar to that observed in other rhabdoviruses [3, 15] (Fig. 1a), allowing prediction of the 3′ and 5′ ends of each mRNA and intergenic sequences between the ORFs. A consensus intergenic sequence can be inferred, 3′-AUUCUUUUU/G(G/A)N n /CUC-5′, which is similar to that observed for other plant rhabdoviruses [3] (Fig. 1b).

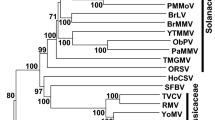
a) Similarity between sequences corresponding to predicted 3′ ends of CBDaV mRNAs, intergenic sequences (IS) and 5′ end of the next mRNA. The sequences are provided in the 3′-5′ sense of the viral mRNAs. Dashes indicate gaps introduced to optimise the alignment, while conserved nucleotides are highlighted in grey. b) Consensus sequences of the gene junction regions of CBDaV compared with other plant rhabdoviruses [3, 9]. c) Maximum-likelihood analysis using the GTR + G + I model, of plant rhabdovirus L polymerase open reading frame nucleotide sequences. The tree is rooted at the midpoint, the insect vectors for each virus are shown, and nucleorhabdovirus and cytorhabdovirus clades are indicated by brackets. Bootstrap values greater than 50 are shown for the major nodes, and the scale indicates the number of substitutions per site. Virus abbreviations are as noted in the text
Based on phylogenetic analysis of the nucleotide and amino acid sequences of the genome and each ORF, CBDaV appears to be a cytorhabdovirus, with NCMV and BYSMV its closest relatives. A phylogenetic tree for the L gene is shown as an example (Fig. 1c). The cytorhabdoviruses form two clades, one containing viruses vectored by planthoppers which includes CBDaV, while the other includes viruses vectored by aphids. CBDV is known to be vectored by the planthopper Tarophagus proserpina. Nucleorhabdoviruses were also separated by their insect vector, suggesting that this could be a useful feature for plant rhabdovirus classification. One striking difference between the planthopper-vectored cytorhabdoviruses is that while NCMV and BYSMV both have multiple ORFs between the P and M coding sequences, CBDaV has a single ORF in this position, similar to the aphid-vectored cytorhabdoviruses. Interestingly, CBDaV has the smallest cytorhabdovirus genome reported to date (Supplementary Table 1), while the genome of TaVCV [9], which has the same genome organisation as CBDaV, is the smallest nucleorhabdovirus genome.
The sequence of the CBDaV genome will now enable investigations into genome variability and virus distribution. Isolation and analysis of CBDaV isolates from bobone- and alomae-diseased taro plants in Solomon Islands and Papua New Guinea will help establish if CBDaV is in fact CBDV and may also shed light on the possible role of CBDV in the etiology of bobone and alomae diseases.
References
Gollifer D, Brown J (1972) Virus diseases of Colocasia esculenta in the British Solomon Islands. Plant Disease Reporter 56:597–599
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Heim F, Lot H, Delecolle B, Bassler A, Krczal G, Wetzel T (2008) Complete nucleotide sequence of a putative new cytorhabdovirus infecting lettuce. Arch Virol 153:81–92
James M, Kenten RH, Woods RD (1973) Virus like particles associated with two diseases of Colocasia esculenta (L.) Schott in the Solomon Islands. J Gen Virol 21:145–153
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP (2009) The sequence alignment/Map format and SAMtools. Bioinformatics 25:2078–2079
Pearson MN, Jackson GVH, Saelea J, Morar SG (1999) Evidence for two rhabdoviruses in taro (Colocasia esculenta) in the Pacific region. Aust Plant Pathol 28:248–253
Revill P, Trinh X, Dale J, Harding R (2005) Taro vein chlorosis virus: characterization and variability of a new nucleorhabdovirus. J of Gen Virol 86:491–499
Revill PA, Jackson GVH, Hafner GJ, Yang I, Maino MK, Dowling ML, Devitt LC, Dale JL, Harding RM (2005) Incidence and distribution of viruses of Taro (Colocasia esculenta) in Pacific Island countries. Aust Plant Pathol 34:327–331
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tanno F, Nakatsu A, Toriyama S, Kojima M (2000) Complete nucleotide sequence of Northern cereal mosaic virus and its genome organization. Arch Virol 145:1373–1384
Tsai CW, Redinbaugh MG, Willie KJ, Reed S, Goodin M, Hogenhout SA (2005) Complete genome sequence and in planta subcellular localization of maize fine streak virus proteins. J Virol 79:5304–5314
Wetzel T, Dietzgen RG, Dale JL (1994) Genomic organization of lettuce necrotic yellows rhabdovirus. Virology 200:401–412
Yan T, Zhu JR, Di D, Gao Q, Zhang Y, Zhang A, Yan C, Miao H, Wang XB (2015) Characterization of the complete genome of Barley yellow striate mosaic virus reveals a nested gene encoding a small hydrophobic protein. Virology 478:112–122
Acknowledgments
Funding support by the Faculty of Health and Environmental Sciences, AUT, is gratefully acknowledged by CH as well as the assistance of Mr. Christopher Puliuvea and Ms. Cintya Del Rio. N.B. is a member of the Argentine Research Council (CONICET) and was supported by a postdoctoral fellowship from INTA, Argentina. The financial assistance of the Australian Centre for International Agricultural Research is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Higgins, C.M., Bejerman, N., Li, M. et al. Complete genome sequence of Colocasia bobone disease-associated virus, a putative cytorhabdovirus infecting taro. Arch Virol 161, 745–748 (2016). https://doi.org/10.1007/s00705-015-2713-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-015-2713-7