
Abstract
Aglaonema bacilliform virus (ABV), a member of the genus Badnavirus in the family Caulimoviridae, is associated with leaf deformation and chlorosis in Aglaonema modestum. The complete genome sequence of a Minnesota isolate of ABV was determined. The ABV genome is 7,178 bp in length and similar in size and organization to those of the members of the genus Badnavirus, containing three open reading frames (ORFs) with the potential to encode three proteins of 14.92, 13.33 and 207.95 kDa, respectively. The last ORF (ORF3) encodes a putative polyprotein with conserved domains, including zinc finger, aspartic protease, reverse transcriptase (RT) and RNase H domains, in that order. Phylogenetic analysis using the amino acid sequence of the ORF3 polyprotein showed that ABV clusters with several isolates of taro bacilliform CH virus (TaBCHV). Pairwise alignment using the highly conserved RT/RNase H region reveals that ABV has the highest level of identity (71%) at the nucleotide level to a Hawaiian isolate of TaBCHV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Aglaonema bacilliform virus (ABV) is a member of the genus Badnavirus (family Caulimoviridae). ABV is associated with leaf mottling and chlorosis of Aglaonema modestum, which occurs widely in commercial production. However, the role of ABV infection in disease expression has not been determined [1]. In previous experiments with the citrus mealybug Planococcus citri, ABV was not transmitted from infected Aglaonema modestum to any of seven other members of the family Araceae, including members of the genera Anthurium, Caladium, Colocasia, Dieffenbachia, Epipremnum, Nephthytis and Philodendron [1]. In this report, the complete genome sequence of ABV and its phylogenetic relationship to other members of the genus Badnavirus is presented.
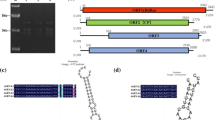
Aglaonema modestum plants showing virus-like symptoms were obtained from commercial greenhouses in Minnesota (USA). Viral particles were isolated from samples showing leaf mottling and premature senescence, and purification was done as described for piper yellow mottle virus (PYMoV) and commelina yellow mottle virus (CoYMV) [2, 3]. Viral particles were examined using transmission electron microscopy (TEM), using samples that were mounted on carbon-coated formvar grids and negatively stained with 2% phosphotungstic acid, pH 7.0 (PTA). A total of 100 virus particles were measured and found to have average dimensions of 30 nm width and 120 nm length (Fig. 1B). No other virus-like particles were detected by electron microscopy in partially purified extracts in symptomatic plants, and further work is required to determine the role of ABV in symptom expression.

A. Aglaonema modestum plants showing symptoms associated with ABV infection. B. Viral particles observed using transmission electron microscopy (TEM). C. Graphic representation of the genome organization of aglaonema bacilliform virus (ABV); orange boxes indicate the predicted open reading frames (ORFs). Conserved domains are denoted by overlying blue boxes
Genomic DNA was extracted from a purified virus suspension and used for sequencing [2, 3]. Briefly, the virus suspension was treated with DNase I (4 units) and RNase (25 μg/mL) at 37 °C for 30 min, followed by digestion with proteinase K (200 μg/mL) in the presence of 1% SDS at 50 °C for 30 min. The mixture was subjected to phenol:chloroform extraction, and nucleic acids were precipitated using ethanol. The extracted DNA was digested in three separate reactions with the restriction enzyme SacI, PstI, or XbaI, and the digestion products were resolved in a 1% agarose gel and stained with 0.5 μg of ethidium bromide pro mL. A single restriction fragment of ~ 7 kb was observed after SacI digestion. Restriction digestion products obtained after SacI digestion of viral genomic DNA were ligated into the SacI site of pBluescript II KS (+), and two clones were sequenced by primer walking. The sequences were assembled using Geneious Prime®. An additional PCR reaction was performed with primers designed to amplify a ~ 1-kb region containing the SacI restriction site. Three of the PCR amplicons were cloned and sequenced. The resulting amplicon sequences matched the sequence of the full-length clone, confirming the completeness of the sequence.
The viral genome consists of a circular dsDNA molecule with a length of 7,718 bp and 40% G + C content. The complete sequence was deposited in the NCBI GenBank database under accession number MH384837. A sequence complementary to the 3’ end of methionine tRNA (5’-TGG TAT CAG AGC AAG GTT-3’) was identified as the primer-binding site for first-strand DNA synthesis and was set as the starting point for the annotation of the circular genomic sequence. An ORF search was done using ORFfinder (NCBI), and a search for conserved domains was done using the NCBI conserved domain database. The genome contains three overlapping ORFs arranged in a fashion similar to that found in other badnaviruses (Fig. 1C) with overlapping stop/start codons between ORF1 and ORF2 (ATGA) and between ORF2 and ORF3 (ATGA) and a 1,021-bp intergenic region. ORF1 (position 400 to 774) encodes a putative protein of approximately 14.92 kDa, with a domain of unknown function (DUF1319) that is restricted to members of the genus Badnavirus. ORF 2 (position 771 to 1136) encodes a predicted protein of 13.33 kDa with no conserved domains. ORF3 (position 1133 to 6556) encodes a putative polyprotein of 207.95 kDa, with conserved domains, including zinc finger, aspartic protease, reverse transcriptase (RT) and RNase H domains. The highest identity values at the amino acid level in ORF 1 (77%), ORF 2 (51%) and ORF 3 (62%) were to their counterparts in taro bacilliform CH virus isolate Et17 from East Africa (MG017324). Pairwise alignments using the RT + RNase H region showed the highest identity (71%) at the nucleotide level to an isolate of TaBCHV from Hawaii, USA (KY359389).
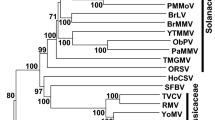
Phylogenetic relationships between ABV and members of the genus Badnavirus were estimated using genetic information from 31 members of this genus, with rice tungro bacilliform virus (X57924) (genus Tungrovirus) as an outgroup. A phylogenetic tree was constructed using the predicted amino acid sequence of the ORF3 putative polyprotein (Fig. 2). Alignment of the amino acid sequences was done using the MAFFT algorithm implemented in Geneious Prime ® [4]. The tree was generated by the maximum-likelihood (ML) method using the RAxML-HPC BlackBox tool from the CIPRES Science Gateway V 3.3 [5]. The ML tree placed ABV in a clade with several isolates of taro bacilliform CH virus (TaBCHV). Based on the phylogenetic reconstruction, ABV and TaBCHV share a common ancestor with piper yellow mottle virus (KJ873042) and several dioscorea bacilliform virus isolates.

Maximum-likelihood (ML) phylogenetic tree of selected members of the genus Badnavirus. The ML tree was constructed using the amino acid sequence encoded by the ORF 3 (polyprotein). Statistical significance was evaluated by bootstrap analysis (1,000 replicates). Bootstrap values higher than 60% are shown at each node. The tree is drawn to scale, with branch lengths expressed in number of substitutions per site. The arrow indicates the position of ABV
References
Lockhart B (1996) Association of a Badnavirus with Premature Leaf Senescence in Aglaonema [abstract], in: abstracts of papers presented at the ninth international symposium on virus diseases of ornamental plants. Phytoparasitica 24:333–334. https://doi.org/10.1007/bf02981414
Lockhart BEL (1990) Evidence for a double-stranded circular DNA genome in a second group of plant viruses. Phytopathology 80:127–131. https://doi.org/10.1094/Phyto-80-127
Lockhart BEL, Kiratiya-Angul K, Jones P et al (1997) Identification of Piper yellow mottle virus, a mealybug-transmitted badnavirus infecting Piper spp. in Southeast Asia. Eur J Plant Pathol 103:303–311. https://doi.org/10.1023/A:1008699414536
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES science gateway for inference of large phylogenetic trees. In: Gateway computing environments workshop (GCE). New Orleans, LA, pp 1–8. https://doi.org/10.1109/gce.2010.5676129
Funding
University of Minnesota, Twin Cities. Alvarez-Quinto R.A is supported by the Republic of Ecuador through the Ecuadorean Science and Technology Secretariat (SENESCYT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare they have no conflict of interest.
Additional information
Handling Editor: Jesus Navas-Castillo.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Alvarez-Quinto, R.A., Lockhart, B.E.L., Moreno-Martinez, J.M. et al. Complete genome sequence of aglaonema bacilliform virus (ABV). Arch Virol 165, 237–239 (2020). https://doi.org/10.1007/s00705-019-04445-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04445-0