
Abstract
Members of the genus Curtovirus (family Geminiviridae) are important pathogens of many wild and cultivated plant species. Until recently, relatively few full curtovirus genomes have been characterised. However, with the 19 full genome sequences now available in public databases, we revisit the proposed curtovirus species and strain classification criteria. Using pairwise identities coupled with phylogenetic evidence, revised species and strain demarcation guidelines have been instituted. Specifically, we have established 77 % genome-wide pairwise identity as a species demarcation threshold and 94 % genome-wide pairwise identity as a strain demarcation threshold. Hence, whereas curtovirus sequences with >77 % genome-wide pairwise identity would be classified as belonging to the same species, those sharing >94 % identity would be classified as belonging to the same strain. We provide step-by-step guidelines to facilitate the classification of newly discovered curtovirus full genome sequences and a set of defined criteria for naming new species and strains. The revision yields three curtovirus species: Beet curly top virus (BCTV), Spinach severe surly top virus (SpSCTV) and Horseradish curly top virus (HrCTV).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The family Geminiviridae comprises seven genera of plant-infecting viruses with circular single-stranded DNA (ssDNA) genomes that are encapsidated within geminate virions. Viral isolates belonging to the genus Curtovirus of the Geminiviridae are transmitted by the leafhopper Circulifer tenellus Baker. Curtoviruses have monopartite genomes of ~2.8–3 kb in length that encode up to seven genes. Three of these are arranged in the virion sense and include the coat protein gene (cp, V1), a regulatory gene (reg, V2) and a movement protein gene (mp, V3). Four are arranged in the complementary sense orientation and include a replication-associated protein gene (rep, C1), a gene expressing a protein that has silencing suppressor functions (ss, C2) [3], a replication enhancer gene (ren, C3) and a symptom determinant gene (sd, C4) [34, 35].
Symptoms in sugar beet plants that have been attributed to curtoviruses were first described in the late 1800s [5, 29], and the documented hosts of viruses in this genus now include more than 300 species of dicotyledonous plants [6, 29, 30]. The known geographical range of the curtoviruses includes the Mediterranean region, the Middle East, the Indian subcontinent and North and Central America.
Over the past 20 years, various guidelines have been proposed by the Geminiviridae Study Group of the International Committee on Taxonomy of Viruses (ICTV) for the nomenclature and classification of geminiviruses [9, 13, 14, 25]. For the genera Begomovirus and Mastrevirus—for which over 1000 full-length genomes have been sampled and sequenced—the most up-to-date variant, strain and species classification guidelines have been based on patterns of pairwise sequence identity displayed by large numbers of analysed genomes coupled with phylogenetic analyses. For example, with respect to pairwise identities calculated between all known mastrevirus genomes, very few pairs of sequences share between 70 and 78 % sequence identity, whereas large numbers of known sequence pairs share either <70 % identity or >80 % identity. This observation has led to the recommendation that all mastrevirus full genome sequences sharing >78 % identity should be classified as belonging to the same species [25].
Up until recently there have been far too few available curtovirus full genome sequences to rationally formulate similar pairwise identity-based guidelines for the classification of virus species in this genus. As a result, the guidelines laid out in 2012 in the 9th Report of the International Committee on Taxonomy of Viruses for curtovirus classification [9] were based largely on the classification scheme proposed for viruses in the genus Begomovirus [14] and proposed that:
-
1)
Viral isolates that share >89 % genome-wide pairwise identity should be classified as belonging to the same species.
-
2)
Viral isolates that have replication-associated proteins (Rep) that are unable to transreplicate one another should be classified as members of different species.
-
3)
Viruses that are serologically distinct may be classified as members of different species or strains.
-
4)
Viruses that have different natural host ranges and symptom phenotypes could be classified as belonging to different strains.
In practice, however, the proposal of new curtovirus species has relied exclusively on the 89 % species demarcation threshold, and as members of new species have been discovered (i.e., new full genome sequences sharing less than 89 % pairwise genome sequence identity with any previously classified full-length curtovirus genome), these have been named based on the hosts from which they were first isolated, the severity of infection symptoms, and symptom phenotypes observed in the field.
However, as has been highlighted in a recent proposal for the classification of mastreviruses [25], pairwise-identity-based classification schemes can in practice yield a large number of conflicting species determinations. This is primarily because there are various ways in which pairwise identities can be calculated, and identity values calculated by different methods can vary widely. The major factors contributing to these discrepancies are:
-
1)
Variations in the nucleotide sequence alignment methods that are used
-
2)
Variations in the gap open and gap extension penalty settings that are applied during alignment
-
3)
Uncertainty over whether pairwise similarities should be calculated within the context of multiple sequence alignments or pairwise alignments
-
4)
Uncertainty over whether “gap” characters introduced during alignment (either in the context of multiple or pairwise sequence alignment) should be ignored or counted as a fifth character state
Nonetheless, pairwise-identity-based viral classification schemes have proven to be highly useful and popular amongst viral taxonomists—especially those working with small viral genomes. Amongst geminivirologists, genome-wide pairwise-identity-based classification approaches have been almost universally adopted and are now well established. Being aware of both the benefits and the potential pitfalls of such approaches, we recently devised a pairwise-identity-based approach for the classification of mastreviruses that effectively eliminates many of the identity calculation discrepancies that are likely to arise during the application of pairwise-identity-based classification guidelines [25]. Our approach, which is implemented in the computer program SDT (available from http://web.cbio.uct.ac.za/SDT), is similar in many respects to the pairwise sequence comparison (PASC) method devised by Bao et al. [4], as it relies on robust and highly reproducible pairwise sequence alignments with complete exclusion of sites with gap characters from the pairwise identity calculations.
Here, we apply the same pairwise-alignment-based identity calculation approach that is currently recommended for mastrevirus classification to the curtoviruses. For this, we first use the distribution of pairwise identity scores between all of the curtovirus genomes that were available in public databases on October 8, 2013, to rationally select new curtovirus species and strain demarcation thresholds, and then apply these thresholds to the re-classification of viruses belonging to this genus.
Rational curtovirus species and strain demarcation criteria
We performed pairwise alignments of the 19 available curtovirus complete genomes and calculated 351 pairwise identity scores for these using the MUSCLE [11, 12], MAFFT [20] and ClustalW [37, 38] alignment approaches implemented in SDT. As was observed in previous analyses carried out on mastreviruses [25], the MUSCLE alignment method yielded the most conservative pairwise identity estimates (Fig. 1), and we therefore focused on the analysis of pairwise alignments yielded by this method for the remainder of our analyses.

New curtovirus species and strain demarcation criteria. a Distribution of pairwise identities of curtovirus full genomes calculated by SDT based on pairwise sequence alignments produced using the MUSCLE, MAFFT and ClustalW alignment programs. b An example of the only conflict encountered using the new species demarcation criterion: BCTV-PeCT and BCTV-CA/Logan
The distribution of pairwise similarity scores calculated from pairwise alignments produced by MUSCLE has notable peaks in the pairwise identity ranges ~66-70 %, 78-89 %, 91-93 % and 96-100 % and “valleys” in the pairwise identity ranges ~71-77 %, and 94-95 % and at 90 % pairwise identity (red line in Fig. 1a). This suggests that a curtovirus species demarcation cutoff could rationally be placed either between 74 and 77 % or at 90 % similarity. Whereas placing the species demarcation threshold within one of the “valleys” would yield a classification scheme with minimal conflict (i.e., the fewest possible instances where the same sequence could justifiably be assigned to two or more different species), placing it at one of the peaks would yield a classification scheme with maximal conflict. Similarly, 90 % or 94 % could be chosen as low-conflict curtovirus strain demarcation thresholds.
Herein, we establish a revision of the existing guidelines that uses the improved similarity calculation approach described above and amends the curtovirus species and strain demarcation thresholds. We establish:
-
(1)
77 % genome-wide pairwise identity as a species demarcation threshold. Therefore, pairs of genomes with >77 % pairwise identity calculated using pairwise MUSCLE alignments with similarities calculated ignoring sites with gaps (such as is implemented in SDT v1.0) should be considered members of the same species.
-
(2)
94 % pairwise identity as a strain demarcation threshold. This, rather than the potential 90 % threshold would allow for the maintenance of the historical strain/variant nomenclature. We therefore propose that pairs of genomes with >94 % pairwise identity as calculated using the same MUSCLE-based pairwise-alignment-based approach as that outlined here should be considered members/variants of the same strain.
Application of the new classification criteria
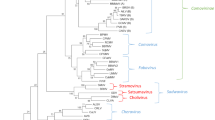
These classification criteria were applied to the 19 curtovirus genomes, and following the construction of a neighbour-joining phylogenetic tree (with the Jukes-Cantor model of nucleotide substitution) with these sequences (Fig. 2), we confirmed that there is strong phylogenetic support for all the curtovirus species and strain groupings that are suggested by these criteria.

Neighbor-joining phylogenetic tree depicting the relationships of known curtovirus full genome sequences (rooted with begomovirus sequences) inferred using the Jukes-Cantor nucleotide substitution model with 1000 bootstrap replicates (branches with less that 75 % support have been collapsed) and a two-dimensional pairwise identity colour matrix with pairwise identities calculated using SDT v1.0 [25]
In accordance with these criteria, we have proposed new names for the 19 known full curtovirus genomes (Table 1). We suggest that the names of these isolates and the names of any curtovirus isolates discovered in the future should have the following form:
<virus species name > - < strain name > [< country/territory code > -<lab codes/old names/host species of origin/sample number/location of origin > -<year of sampling >]
Virus species name
This is the ICTV-accepted name (or acronym thereof) of a group of viruses sharing >77 % genome-wide pairwise sequence identity. If the sequence has <77 % genome-wide pairwise identity to all curtovirus sequences previously classified as belonging to any recognised species, the virus should be considered a member of a new species, and a unique name should be assigned (a name that is not currently in use for any ICTV-recognised species). This name should be based on both the name of the host from which the virus was originally isolated and the type of symptoms produced in this host. Further, we recommend that country, city, town, village or province names not be used in naming new viral species (e.g., Spinach curly top Arizona virus), as this adds to downstream complications when similar sequences are found in other territories/countries/regions.
Strain name
In order to maintain some historical nomenclature, the following as strain identifiers are retained: Worland (Wor), Mild (Mld), Severe (Svr), California/Logan (CA/Logan), pepper curly top (PeCT), pepper yellow dwarf (PeYD), spinach curly top (SpCT) and Colorado (CO). Further, we propose that new strains that are identified in the future follow a nomenclature that is consistent with observable biological differences between the members of the same strain. When sufficient knowledge is available, it is strongly recommended that the strain descriptor genuinely reflect the characteristics of all members of a strain. For example, if it is established that multiple different isolates of a BCTV strain are especially well adapted relative to other BCTV strains to infecting a particular host such as spinach, then it would be entirely justifiable to name the strain BCTV-spinach. In the future, symptom severity descriptors (such as mild or severe) should only be used as strain names when such phenotypes are observed in multiple genetically distinct members of a strain.
Isolate descriptor
Within the square brackets (“[ ]”) the isolate descriptor may contain any number of sub-fields separated by hyphens (“-”). Whereas the first sub-field should always be the two-letter international code of the country/territory in which the isolate was last present in the field (Supplementary table 1), the last sub-field should always be the year in which the isolate was last present within living tissue. If the year in which the isolate was removed from the field differs from the date on which it was last present within living tissue (as is sometimes the case when isolates are propagated in the laboratory), then the date when the virus was removed from the field should be included in one of the internal sub-fields. Between the first and last sub-fields any additional short useful descriptors can be placed (for example, the laboratory or field identification codes of the samples from which the isolate was obtained, the city where the sample was found and the host species from which the virus was isolated). Please note that although the Ninth ICTV Report’s recommendations for geminivirus nomenclature [9] suggested the use of “:” to separate the sub-fields in the isolate descriptor, this symbol can cause problems in various phylogenetic tree drawing programs, which, when reading phylogenetic trees in Newick format, will misinterpret numbers after the “:” symbol as representing branch length information.
Resolving conflicts that may arise within the new curtovirus classification system
We have specifically recommended curtovirus species and strain demarcation thresholds that minimise ambiguous species and strain classifications amongst the currently available curtovirus sequences. However, as new curtovirus genomes are sequenced and classified under this system it is probable that in some cases conflict will arise as a result of:
-
1.
An isolate having >77 % genome-wide pairwise identity to isolates that have been assigned to two different species.
-
2.
An isolate having >77 % genome-wide pairwise identity to one or a few isolates assigned to a particular species, even though it shares <77 % identity with the majority of characterised isolates in that species.
-
3.
An isolate having >94 % genome-wide pairwise identity to isolates that have been assigned to two different strains.
-
4.
An isolate having >94 % genome-wide pairwise identity to one or a few isolates assigned to a particular strain, even though it shares <94 % identity with the majority of characterised isolates in that strain.
We recommend that the following steps be taken to resolve such conflicts:
-
1.
The new isolate should be considered to belong to the species containing the isolate with which it shares the highest percentage genome-wide pairwise identity.
-
2.
The new isolate should be classified as belonging to any species in which it shares >77 % genome-wide pairwise identity with any one isolate previously classified as belonging to that species, even if it has <77 % genome-wide pairwise identity to all other isolates classified as belonging to that species.
-
3.
The new isolate should be considered to belong to the strain containing the isolate with which it shares the highest degree of identity.
-
4.
The new isolate should be classified as belonging to any strain in which it shares >94 % identity with any one isolate previously classified as belonging to that strain, even if it is <94 % identical to all other isolates classified as belonging to that strain.
That the proposed >77 % species demarcation threshold yields a single type 2 conflict (Figs. 1b and 2) emphasises the importance of having clear guidelines for resolving such conflicts. Specifically, although the BCTV-PeCT and BCTV-CA/Logan isolates share between 75 and 76 % genome-wide pairwise identity, they both share >78 % genome-wide pairwise identity to all other BCTV isolates (Fig. 2), and therefore, based on the recommended approach for conflict resolution, both would be classified as strains of BCTV. Furthermore, our recommendation can be complemented with biological properties exhibited during viral infection.
A step-by-step guide to classifying a new curtovirus full genome sequences
-
(1)
A ‘nucleotide BLAST’ analysis (accessible via http://blast.ncbi.nlm.nih.gov/Blast.cgi) of the NCBI ‘Nucleotide collection’ database should be performed to identify the species whose members have sequences most similar to the new sequence. The nucleotide database at the NCBI website (http://www.ncbi.nlm.nih.gov/nuccore/) can also be searched using the search term “txid10813[Organism:exp] AND 2500:4000[SLEN]”, which will return all curtovirus nucleotide sequences that are between 2500 and 4000 nucleotides long.
-
(2)
The new sequence should be added to the set of sequences obtained from the NCBI BLAST or NCBI nucleotide database websites and should be saved in FASTA format (see supplementary material).
-
(3)
Regardless of how datasets are compiled, sub-genome-length sequences should ultimately be removed from FASTA files that are intended for use in pairwise sequence identity analyses.
-
(4)
Prior to any analysis check, and if need be, ensure that all the sequences being analysed start at the same genomic coordinate (ideally at the nicking site within the conserved nonanucleotide at the origin of replication).
-
(5)
Use the MUSCLE option in SDT v1.0 (available at http://web.cbio.uct.ac.za/SDT) or any other program that uses the MUSCLE alignment algorithm (with pairwise deletion of gaps) to calculate identities between every pair of sequences in the dataset. If using SDT, these pairwise identities should be saved either as a column or matrix csv format that can then be opened in a spreadsheet program such as Microsoft Excel. Most other software will have similar options to SDT to generate csv files of pairwise identities.
-
(6)
If the sequence shares <77 % genome-wide pairwise identity to any other known curtovirus sequence, then an appropriate species name should be proposed (see above for guidelines on doing so).
-
(7)
If the sequence shares <94 % genome-wide pairwise identity to all isolates described for that species, then a strain name should be proposed.
Recombination in curtoviruses
As recombination has played a major role in the evolution of geminiviruses, we analysed the 19 curtovirus genomes using the RDP, GENECONV [26], Bootscan [23], Maxchi [31], Chimera [27], Siscan [15] and 3Seq [7] methods implemented in RDP4 [24]. Only potential recombination events that were detected with three or more of these seven methods (with associated p-values of <0.05) coupled with phylogenetic support for recombination having occurred were accepted as evidence of genuine recombination events.
The 18 recombination events thus detected are summarised in Fig. 3. It is interesting to note that the genomes of BCTV-Svr and BCTV-PeCT display evidence of at least four independent recombination events. Also, besides a large number of intra-species recombination events (n = 14) there is also clear evidence of inter-species recombination (events 11, 12, 15, 16). Similar to observations in begomoviruses and mastreviruses, there is potentially a recombination breakpoint hotspot in the region between the 3’ ends of the cp (V1) and rep (C1) genes.

a Maximum-likelihood phylogenetic tree (1000 bootstrap replicates) of curtoviruses (with recombinant regions removed) inferred with PHYML [16] using the TN93+G nucleotide substitution model (determined as fitting the data best by jModelTest [28]). Branches with <75 % bootstrap support have been collapsed. b Cartoon illustrating the regions of curtovirus genomes that have been acquired through recombination. c. Details of recombination events detected in the curtovirus genomes. Methods used to detect recombination are RDP (R) GENCONV (G), BOOTSCAN (B), MAXCHI (M), CHIMERA (C), SISCAN (S) and 3SEQ (T). The method with the most significant associated p-value is indicated in bold for each event
So as to determine the potential impact of recombination on the proposed curtovirus classification system, a maximum-likelihood phylogenetic tree (with PHYML3 [16], TN93+G determined to be the best nucleotide substitution model by jModelTest [28], 1000 bootstrap replicates and the collapsing of branches with <75 % bootstrap support) was constructed using the “non-recombinant” fractions of the curtovirus genomes (represented by uncoloured genome regions in Fig. 3). This tree indicated that, even when accounting for recombination, there is phylogenetic support (75 % bootstrap value on the branch separating the BCTV isolates from HrCTV and SpSCTV) for the existence of the three curtovirus species indicated in the revised classification system.
Conclusions
In the 9th Report of the International Committee on Taxonomy of Viruses [9], the curtovirus classification was based primarily on the 89 % species demarcation criterion previously determined for the genus Begomovirus [14] with secondary considerations being given to the biological properties of viruses, such as host range, symptom phenotype and serology. Based on this classification system, the ICTV recognised seven species within the genus Curtovirus.
In this communication, we have established revised genome-wide pairwise-identity-based species and strain demarcation criteria for the classification of curtoviruses, i.e., curtovirus genome sequences with <77 % genome-wide pairwise identity to all previously classified curtoviruses as calculated using either the SDT-based approach described here or an exactly equivalent method. Similarly, a genome sequence found to share >77 % but <94 % identity to isolates from a previously established curtovirus species using this same approach should be considered to belong to a new strain of that species.
Based on these revised classification criteria, the genus Curtovirus now has only three species: Beet curly top virus, Horseradish curly top virus and Spinach severe curly top virus. The species demarcation criteria we have applied here for curtoviruses and that which we have applied previously to mastreviruses [25] are both predominantly based on pairwise full-genome nucleotide sequence identities coupled with phylogenetic support. This approach has been critiqued by Van Regenmortel et al. [39], who favour the concept of a virus species being a polythetic class of viral isolates constituting a replicating lineage in a particular ecological niche and argue against a proposal by A. King, M. Adams, E. Lefkowitz and E. Carstens (proposal 2011.002sG), which states ‘A species is a monophyletic group of viruses whose properties can be distinguished from other species by multiple criteria’ (the criteria include natural and experimental host ranges, pathogenicity, vector specificity, cell and tissue tropism and degrees of relatedness of their genes and genomes). In the case of viruses with small genomes (where full genome sequences can be easily determined), genome-wide sequence comparisons provide substantially more objective information than can visual observations of biological traits such as symptom phenotype. For example, the 2930-nucleotide genome of BCTV-Wor (U56975) contains 2930 discrete bits of information that can, with total objectivity, indicate whether it is more closely related to other curtoviruses found in beets than it is to those found in horseradish or spinach. In the case of the curtoviruses, the species demarcation criteria that we have proposed suggest the existence among the currently known curtoviruses of only three monophyletic species (Beet curly top virus, Horseradish curly top virus and Spinach severe curly top virus) that are also distinguishable by the host species from which they have been obtained. Therefore, although our genome-sequence-based approach to virus classification is not universally supported [39], it does, in this case at least, yield a completely objective classification system that is consistent with the observed biological properties of curtoviruses.
Finally, it should be noted that while the Executive Committee of the ICTV has approved these genome-wide pairwise-identity based curtovirus species and strain demarcation criteria [1], it has also stressed that new species proposals must be supported by additional phylogenetic evidence.
References
Adams MJ, King AM, Carstens EB (2013) Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2013). Arch Virol 158:2023–2030
Baliji S, Black MC, French R, Stenger DC, Sunter G (2004) Spinach curly top virus: a newly described curtovirus species from Southwest Texas with incongruent gene phylogenies. Phytopathology 94:772–779
Baliji S, Sunter J, Sunter G (2007) Transcriptional analysis of complementary sense genes in Spinach curly top virus and functional role of C2 in pathogenesis. Mol Plant Microbe Interact 20:194–206
Bao Y, Chetvernin V, Tatusova T (2012) PAirwise Sequence Comparison (PASC) and its application in the classification of filoviruses. Viruses 4:1318–1327
Bennett CW, Tanrisever A (1957) Sugarbeet curly top disease in Turkey. Plant Dis Report 41:721–725
Bennett CW (1971) The curly top disease of sugarbeet and other plants Monograph 7. The American Phytopathological Society, St. Paul
Boni MF, Posada D, Feldman MW (2007) An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176:1035–1047
Briddon RW, Stenger DC, Bedford ID, Stanley J, Izadpanah K, Markham PG (1998) Comparison of a beet curly top virus isolate originating from the old world with those from the new world. Europ J Plant Pathol 104:77–84
Brown JK, Fauquet CM, Briddon RW, Zerbini M, Moriones E, Navas-Castillo J (2012) Virus taxonomy: Ninth report of the International Committee on Taxonomy of Viruses. Academic press, London; Waltham
Chen LF, Vivoda E, Gilbertson RL (2011) Genetic diversity in curtoviruses: a highly divergent strain of Beet mild curly top virus associated with an outbreak of curly top disease in pepper in Mexico. Arch Virol 156:547–555
Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform 5:113
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acid Res 32:1792–1797
Fauquet CM, Maxwell DP, Gronenborn B, Stanley J (2000) Revised proposal for naming geminiviruses. Arch Virol 145:1743–1761
Fauquet CM, Briddon RW, Brown JK, Moriones E, Stanley J, Zerbini M, Zhou X (2008) Geminivirus strain demarcation and nomenclature. Arch Virol 153:783–821
Gibbs MJ, Armstrong JS, Gibbs AJ (2000) Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16:573–582
Guindon S, Delsuc F, Dufayard JF, Gascuel O (2009) Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137
Hernandez-Zepeda C, Varsani A, Brown JK (2013) Intergeneric recombination between a new, spinach-infecting curtovirus and a new geminivirus belonging to the genus Becurtovirus: first New World exemplar. Arch Virol 158:2245–2254
Hernandez C, Brown JK (2010) First report of a new Curtovirus species, Spinach severe curly top virus, in Commercial Spinach Plants (Spinacia oleracea) from South-Central Arizona. Plant Dis 94:917
Hormuzdi SG, Bisaro DM (1993) Genetic analysis of beet curly top virus: evidence for three virion sense genes involved in movement and regulation of single- and double-stranded DNA levels. Virology 193:900–909
Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33:511–518
Klute KA, Nadler SA, Stenger DC (1996) Horseradish curly top virus is a distinct subgroup II geminivirus species with rep and C4 genes derived from a subgroup III ancestor. J Gen Virol 77:1369–1378
Lam N, Creamer R, Rascon J, Belfon R (2009) Characterization of a new curtovirus, pepper yellow dwarf virus, from chile pepper and distribution in weed hosts in New Mexico. Arch Virol 154:429–436
Martin DP, Posada D, Crandall KA, Williamson C (2005) A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses 21:98–102
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463
Muhire B, Martin DP, Brown JK, Navas-Castillo J, Moriones E, Zerbini FM, Rivera-Bustamante R, Malathi VG, Briddon RW, Varsani A (2013) A genome-wide pairwise-identity-based proposal for the classification of viruses in the genus Mastrevirus (family Geminiviridae). Arch Virol 158:1411–1424
Padidam M, Sawyer S, Fauquet CM (1999) Possible emergence of new geminiviruses by frequent recombination. Virology 265:218–225
Posada D, Crandall KA (2001) Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc Natl Acad Sci USA 98:13757–13762
Posada D (2008) jModelTest: phylogenetic model averaging. Mol Biol Evol 25:1253–1256
Severin HH (1927) Crops naturally infected with sugar beet curly-top. Science 66:137–138
Severin HHP (1929) Additional host-plants of curly top. Hilgardia 3:595–636
Smith JM (1992) Analyzing the mosaic structure of genes. J Mol Evol 34:126–129
Soto MJ, Gilbertson RL (2003) Distribution and rate of movement of the curtovirus Beet mild curly top virus (Family geminiviridae) in the beet leafhopper. Phytopathology 93:478–484
Stanley J, Markham PG, Callis RJ, Pinner MS (1986) The nucleotide sequence of an infectious clone of the geminivirus beet curly top virus. EMBO J 5:1761–1767
Stenger DC, Carbonaro D, Duffus JE (1990) Genomic characterization of phenotypic variants of beet curly top virus. J Gen Virol 71:2211–2215
Stenger DC (1994) Complete nucleotide sequence of the hypervirulent CFH strain of beet curly top virus. Mol Plant Microbe Interact 7:154–157
Sunter G, Sunter JL, Bisaro DM (2001) Plants expressing tomato golden mosaic virus AL2 or beet curly top virus L2 transgenes show enhanced susceptibility to infection by DNA and RNA viruses. Virology 285:59–70
Thompson JD, Plewniak F, Poch O (1999) A comprehensive comparison of multiple sequence alignment programs. Nucleic Acids Res 27:2682–2690
Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. In: Andreas D Baxevanis et al., Current protocols in bioinformatics / editoral board, Chapter 2:Unit 2 3
Van Regenmortel MH, Ackermann HW, Calisher CH, Dietzgen RG, Horzinek MC, Keil GM, Mahy BW, Martelli GP, Murphy FA, Pringle C, Rima BK, Skern T, Vetten HJ, Weaver SC (2013) Virus species polemics: 14 senior virologists oppose a proposed change to the ICTV definition of virus species. Arch Virol 158:1115–1119
Velasquez-Valle R, Mena-Covarrubias J, Reveles-Torres LR, Arguello-Astorga GR, Salas-Luevano MA, Mauricio-Castillo JA (2012) First report of Beet mild curly top virus in Dry Bean in Zacatecas, Mexico. Plant Dis 96:771–772
Acknowledgements
JNC and EM are members of the Research Group AGR-214, partially funded by Consejería de Economía, Innovación y Ciencia, Junta de Andalucía, Spain, cofinanced by FEDER-FSE. AV and DPM are supported by the National Research Foundation, South Africa.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
705_2014_1982_MOESM1_ESM.fas
FASTA file of curtoviruses sequences (full genomes) available in public databases (downloaded on October 8, 2013) (FAS 57 kb)
Rights and permissions
About this article
Cite this article
Varsani, A., Martin, D.P., Navas-Castillo, J. et al. Revisiting the classification of curtoviruses based on genome-wide pairwise identity. Arch Virol 159, 1873–1882 (2014). https://doi.org/10.1007/s00705-014-1982-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-014-1982-x