
Abstract
This study reports the molecular epidemiology and genetic characterization of human respiratory syncytial virus (RSV) samples collected in Thailand from January 2010 to December 2011. In total, 1,315 clinical samples were collected from Bangkok and Khon Kaen provinces and were screened by semi-nested PCR for RSV infection. We found 74 samples (27.7 %) and 71 samples (6.8 %) to be RSV positive for Bangkok and Khon Kaen, respectively, and we sequenced 122 of these samples. Phylogenetic analysis revealed that 100 of the RSV-A-positive samples clustered into either genotype NA1 or the recently discovered genotype ON1 strain, which has a 72-nucleotide duplication in the second variable region of its G protein. Moreover, 22 of the RSV-B-positive samples clustered into four genotypes; BA4, BA9, BA10 and genotype THB, first described here. The NA1 genotype was found to be the predominant strain in 2010 and 2011. The ON1 strain detected in this study first emerged in 2011 and is genetically similar to ON1 strains characterized in other counties. We also describe the THB genotype, which was first identified in 2005 and is genetically similar to the GB2 genotype. In conclusion, this study indicates the importance of molecular epidemiology and characterization of RSV in Thailand in order to better understand this virus. Further studies should be conducted to bolster the development of antiviral agents and a vaccine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human respiratory syncytial virus (RSV) is an important contributor to lower respiratory tract infections (LRTIs) in young children and infants across the world [1]. The prevalence of the virus is highest during rainy seasons in tropical zones and during the winter in temperate zones [2]. The clinical characteristics of patients with RSV infection are various and include manifestations in the form of bronchitis, bronchiolitis and pneumonia. In addition, some children display fever, cough, rhinorrhea, wheezing, dyspnea and hypoxia [3], and they are more likely to develop a severe infection if they have an underlying condition, such as chronic pulmonary disease, premature birth or congenital heart disease [4]. To date, neither vaccines nor effective treatment for this virus have been developed [5].
RSV is classified as a member of the family Paramyxoviridae, genus Pneumovirus. Its genome is comprised of negative-sense, single-stranded RNA, which is translated into eleven distinct viral proteins. The membrane of the virus contains two major surface glycoproteins; the attachment (G) and the fusion (F) proteins. The G protein is associated with the attachment of viral particles to a host-cell membrane, while the F protein’s function is to fuse the viral membrane to the host membrane and to also fuse adjacent cells, which contributes to syncytium formation [6, 7].
Strains of this virus can be divided into two main groups by serological reaction between glycoproteins and monoclonal antibodies: RSV group A (RSV-A) and RSV group B (RSV-B) [8, 9]. These groups in turn can be further identified by genetic classification of the G protein [10, 11]. RSV-A consists of eleven genotypes, GA1 to GA7 [12, 13], SAA1 (South African strain) [14], NA1 and NA2 from Niigata, Japan [15] and ON1 from Ontario, Canada [16] while RSV-B is classified into twenty genotypes, GB1 to GB4 [12], SAB1 to SAB4 [14, 17], URU1, URU2 [18] and BA1 to BA10 (Buenos Aires strain). There can be, however, significant similarities between disparate strains. For example, BA strains have a 60-nucleotide duplication located in the second variable region of the G protein [19–21]. This is similar to the ON1strains of RSV-A, which have a 72-nucleotide duplication in the same region [16].
The G protein is a member of the glycoprotein type II family and has a unique nucleotide sequence and structure compared to those of other paramyxoviruses [22]. The G protein contains three major domains: the cytoplasmic domain and transmembrane domain, both of which are highly conserved, and the ectodomain. The ectodomain contains two variable regions split by a sequence of 13 conserved amino acids [23–25], and a second variable region, located in the C-terminal portion of the ectodomain, where all of the variations in the G protein are believed to occur. Thus, this region has been particularly helpful in revealing the evolutionary and molecular epidemiology of RSV [12, 13].
While both RSV-A and RSV-B and their various genotypes may contribute to an epidemic, only one genotype is predominant in any given year, and it may be replaced by another genotype in the following year [12, 14]. In addition, the nucleotide sequence of the G gene – especially in the variable regions – tends to change with time within the same genotype. This suggests that selective pressure exerted by the host’s immune system determines genotype selection from season to season [26, 27]. Interestingly, this model of selection is similar to the evolutionary behavior of influenza virus type B [28, 29].
In Thailand, RSV is endemic, and epidemics occur during the rainy season from July to October, with a peak in August or September of each year [30, 31]. Most patients are children below the age of 5 years or elderly [32], and RSV studies in Thailand involving viral reactions with monoclonal antibodies have revealed that the predominant isotype contributing to incidence can vary from year to year [31, 33]. There has been one study on molecular characterization of RSV strains in Thailand, which showed that GA2 of RSV-A and BA4 of RSV-B were the predominant genotypes in 2007 [34]. However, this study observed a short timeframe and utilized a limited number of samples. Thus, further molecular characterization of RSV-A and RSV-B virus in Thailand is still required. The objective of our study was to carry out an epidemiological investigation and molecular characterization of a large number of RSV samples collected throughout Thailand from January 2010 to December 2011.
Materials and methods
The protocol of this study was approved by the Ethics Committee of the hospital and Faculty of Medicine, Chulalongkorn University (IRB 386/53). All stored samples in this retrospective study have been used with permission from the Director of King Chulalongkorn Memorial Hospital. Patient identifiers, including personal information (name, address) and hospitalization number, were removed from these samples to protect patient confidentiality, and they did not appear in any part of this study. IRB waived the requirement for consent because the samples were anonymous.
Clinical samples
The majority of the study specimens were collected by nasopharyngeal aspiration (NPA), nasal swab, and throat swab from patients with acute respiratory tract symptoms, aged 1 day to17 years. The NPA and some nasal swab samples were retrieved from patients diagnosed at the Department of Pediatrics, Chulalongkorn Memorial Hospital, and other hospitals located in Bangkok from July 2010 to December 2011. The throat swab samples were retrieved from patients with acute respiratory tract infections from Chumphae Hospital, Khon Kaen Province, between January 2010 and October 2011. Specimens sent to the Center of Excellence in Clinical Virology, Chulalongkorn University, were kept at −70 °C until laboratory tests were conducted.
RNA extraction and reverse transcription
RNA was extracted from 200 μl of each sample using a HiYieldTM Viral Nucleic Acid Extraction Kit (RBC Bioscience, Taipei, Taiwan) according to the manufacturer’s instructions. RNA was reverse transcribed into cDNA using random hexamer primers and ImProm-IITM Reverse Transcriptase (Promega, Madison, WI) according to the manufacturer’s instructions. Briefly, RNA was incubated with random hexamer primers at 70 °C for 5 min, and the first strand was extended for 2 hours at 42 °C. Reverse transcriptase was then heat inactivated at 70 °C for 15 min.
PCR amplification and DNA sequencing
In this study, we applied semi-nested PCR to differentiate between RSV-A and RSV-B strains. The specific primers we used for the second variable region of the G protein are described here: We used RSVA_227F1 (5′-TACAAGATGCAACAARCCAGATCA-3′, position 227-250 of the G gene of strain A2) and RSVA_1072R (5′-CTAACTGCACTGCATGTTGATTGA-3′, position 122-99 of the F gene) as outer sense and outer anti-sense primers for RSV-A identification, and we used RSVB_225F1 (5′-AGTTCAAACAATAAAAAACCACACTG-3′, position 225-250 of G gene of strain 9320) and RSVB_1009R (5′-TGCATTAATAGCAAGAGTTAGGAAG-3′, position 57-33 of the F gene) for RSV-B identification. The cDNA was amplified using a Perfect TaqPlus Master Mix Kit (5 prime GmbH, Hamburg, Germany) according to the manufacturer’s instructions. PCR conditions consisted of initial denaturation at 94 °C for 3 minutes, followed by 40 cycles of denaturation at 94 °C for 18 s, annealing at 55 °C for 21 s, extension at 72 °C for 1.5 min and a final extension at 70 °C for 10 min. For semi-nested PCR, we utilized the inner forward primer RSVA_234F2 (5′-TGCAACAARCCAGATCAAGAACAC-3′, position 234-257 of the G gene) and RSVB_269F2 (5′-ACCTTACTCAAGTCYCACCAGAAA-3′, position 269-292 of the G gene) for RSV-A and B, respectively. The same reaction conditions as described above were used but reduced to 30 rounds. PCR products were separated by 2 % agarose gel electrophoresis, stained with ethidium bromide, and then visualized under UV light for amplicon detection. Amplicons were purified using an Agarose Gel Extract Mini Kit (5 prime GmbH, Hamburg, Germany) according to the manufacturer’s instruction. The purified PCR products were subjected to direct sequencing of the G gene by First BASE Laboratories Sdn Bhd (Selangor Darul Ehsan, Malaysia).
Phylogenetic tree construction and analysis of partial nucleotide sequences of the G gene
Partial nucleotide sequences of the G genes (about 862 bp for RSV-A and 765 bp for RSV-B) were analyzed using the Basic Local Alignment Search Tool (BLAST) program available on the NCBI homepage (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Nucleotide sequences were edited in comparison with reference strains available in the GenBank database using Chromas Lite software version 2.01 and SeqMan (DNA STAR, Madison, WI). The sequences that represented each genotype were retrieved from GenBank and aligned using ClustalW via BioEdit software version 7.0.9 (http://www.mbio.ncsu.edu/bioedit/bioedit.html). Sequences were analyzed to identify substitutions or insertions in the strains studied. Phylogenetic trees of RSV-A and RSV-B were constructed using the neighbor-joining method in MEGA 4.0.2 with 1,000 bootstrap replicates to identify the genotypes in this study. Pairwise nucleotide distances (p-distance) were calculated to compare the differences between and within genotypes. In addition, SPSS statistics version 17.0 was used to calculate the average age of the study subjects.
Results
Clinical samples
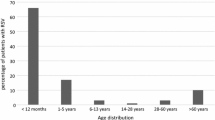
Samples from Bangkok (n = 267) and Khon Kaen (n = 1,048) were collected between 2010 and 2011 and tested for RSV by semi-nested PCR. Seventy-four (27.7 %) and 71 (6.8 %) samples from Bangkok and Khon Kaen, respectively had detectable RSV. The average age of patients with RSV infection from the Bangkok cohort was 2.2 ± 3.1 in 2010 and 1.3 ± 1.2 in 2011. Meanwhile, the average age of patients with RSV infection for the Khon Kaen cohort was 1.5 ± 0.9 in 2010 and 2.2 ± 1.4 in 2011. The majority of RSV-infected patients were infants ≤2 years of age. The male/female ratio was 1.5:1 for Bangkok and 1:1 for Khon Kaen. Among 145 positive samples, 119 were identified as RSV-A, while 26 were RSV-B. Out of those positive samples, 100 of the RSV-A and 22 of the RSV-B samples were sequenced; however, 19 RSV-A samples and 4 RSV-B samples could not be sequenced due to an insufficient quantity of material. RSV sequences obtained in this study were submitted to the GenBank database under accession numbers KC342350- 449 for RSV-A and KC342327 -348 for RSV-B.
Phylogenetic analysis and molecular epidemiology
The phylogenetic tree of 100 RSV-A sequences clustered into two main groups with previously designated genotypes. The major group consisted of 90 (90 %) sequences classified as genotype NA1, while the remaining 10 (10 %) sequences grouped with the recently described genotype ON1. The phylogenetic tree showed that the NA1 genotypes clustered together, with bootstrap values of 83 %. This group was further classified into sub-branches that may be labeled as subtypes [12]. The individual pairwise nucleotide distance (p-distance) between sequences in NA1 from this study was as high as 0.064. As for the ON1 genotype, this genotype is separate from the NA1 genotype and clustered with a bootstrap value of 86 %. The p-distance for this group was 0.029. Many sequences in this study were identical; thus, only representative samples were selected for reconstruction of the phylogenetic tree (Fig. 1).

Phylogenetic tree of RSV-A constructed by the neighbor-joining method using MEGA version 4.0.2. The sequences from this study are indicated by solid triangles, and genotypes are shown in bold letters in brackets. The numbers at each branch node are bootstrap values calculated from 1,000 replicates of reconstruction. Bootstrap values above 50 % are shown, and the tree was unrooted
The 22 sequences of RSV-B clustered into four genotypes: 13 (59.1 %) and 6 (27.3 %) sequences clustered with the recently discovered BA genotypes BA9 and BA10, while one (4.5 %) sequence was determined to be BA4. Although these sequences clustered with previous genotypes, bootstrap values were not supportive of the three clusters. Interestingly, the remaining two sequences clustered with Beijing strains H5598 and H5601 described by Deng and colleagues [35] but were not classified as a separate genotype. The bootstrap value of this cluster was 99 %, and the p-distance between members ranged from 0.004 to 0.041, suggesting that this is a new genotype, as the bootstrap value is greater than 70 % and the p-distance between members of the cluster is less than or equal to 0.07 [14]. The new genotype in this study has been called the Thailand B strain (THB) (Fig. 2). Furthermore, the p-distance between CU2010/5 and GB2 (CH93-9b) was 0.077, while the p-distance between CU2011/81 and GB2 was 0.082.

Phylogenetic tree of RSV-B constructed by the neighbor-joining method using MEGA version 4.0.2. The sequences from this study are indicated by solid triangles, and genotypes are shown in bold letters in brackets. The numbers at each branch node are bootstrap values calculated from 1,000 replicates of reconstruction. Bootstrap values above 50 % are shown, and the tree was unrooted
In this study, RSV infection was initially detected from March 2010 until November 2011. The peak of RSV infection was in September 2010 and October 2011. Interestingly, the number of infected individuals was high in March 2011 even though this lay outside of the period of peak incidence. Furthermore, the results have shown RSV-A to be the predominant subgroup in this study. Molecular analysis showed that genotype NA1 of RSV-A was predominant during the study period, with 31/45 isolates (68.9 %) found in 2010 and 59/77 isolates (76.6 %) found in 2011. At the same time, the circulation of ON1 genotype of RSV-A was only detected in 10 out of 77 isolates (13 %) in 2011. As for RSV-B, the BA9 genotype circulated throughout both years of the study with 7 or 45 (15.6 %) and 6 of 77 (7.8 %) isolates found in 2010 and 2011, respectively. The BA10 genotype was found in 6 of 45 isolates (13.3 %) in 2010, while the BA4 genotype was found in 1 of 77 isolates (1.3 %) in 2011. Finally, the new THB genotype of RSV-B was found in 1 of 45 (2.2 %) and 1 of 77 (1.3 %) patients in 2010 and 2011, respectively.
Amino acid sequence characteristics and N-linked glycosylation site
The stop codon of the second variable region of the Thai RSV-A in this study was at position 298 for genotype NA1 and at position 322 for genotype ON1. In comparison to the RSV-A reference strain A2, the Thai NA1 and ON1 strains had the following amino acid substitutions: S222P/L, P226L, E233K/R, I244R/G, L258H, M262E/K, F265L, S269T, S280Y/H, P286L, P289S/Y, S290P/L, P292S, P293S, P296T/I and R297K/E. There were three unique amino acid substitutions for ON1: E232G, T253K, and S314L. The amino acid sequences of the second variable region of the G protein of the Thai RSV-A isolates revealed four N-linked glycosylation sites. The first site is located at aa 237. This site is conserved among ON1 strains, while in some NA1 strains the substitution of N237D results in the loss of an N-linked glycosylation site. The second and third sites, located at aa 251 and 273, respectively, were found in most of NA1 sequences. The fourth glycosylation site was located at aa 294 for NA1 genotype and at aa position 318 for the ON1 genotype (Fig. 3a). Comparison of the ON1 genotypes in this study to the ON1 genotypes from GenBank (Canada, Japan, South Korea and South Africa) shows that the sequences are conserved except for ON1 from South Africa. This sequence has an E308K substitution, which is not seen in the ON1 strains from other countries (Fig. 3b).

(a) Amino acid sequence alignment of the second variable region of representative Thailand RSV-A strains with prototype strain A2 (GenBank accession number M11486) and (b) alignment of the second variable region of ON1 strains from GenBank and ON1 strains from Thailand. Identical residues are indicated by dots. Stop codons are indicated by asterisks, and N-linked glycosylation sites are indicated by gray shading. For the ON1 strain, the duplicated regions are underlined, and the amino acids that are unique to the South African strain are framed by a rectangle. Genotype names are indicated to the right of the brackets
The RSV-B strains from Thailand had stop codons at aa 296 for CU2010/5 and CU2011/81, at aa position 313 for all BA strains, and at aa 320 for CU2011/205. The amino sequences of the Thai BA strains were aligned and compared to that of the BA prototype BA4128/99B. The alignment showed that S247P was found in all Thai BA strains. Unique amino acid substitutions for strain-specific genotypes were investigated. The substitution of V271A was unique for all Thai BA9 strains. The BA10 strain contained three unique substitutions, K213R, E226D and E292G. In addition, the prototype strain CH18537 was used for comparison with the non-BA-strain, THB, in this study. The comparison showed that R214I, V251M, K258N, E272D and T295V are unique in all THB strains, but D215V, I227A and K234N are unique for the Thai THB genotype only. Three potential N-linked glycosylation sites were identified in the Thai RSV-B strains. The first site is located at aa 258. This site is unique for all THB strains. The second site is located at aa 296 or 276 (non BA), and the third site is at aa 310 or 290 (non BA). Furthermore, the potential glycosylation sites of positions 230 and 253 were found only in strain H5598 of THB and CU2011/195 of BA9, respectively (Fig. 4).

Amino acid sequence alignment of the second variable region of Thailand RSV-B strains with prototype strain CH18537 for non BA strains (GenBank accession number M17213) and BA4128/99B for BA strains (GenBank accession number AY333364). Identical residues are indicated by dots. Stop codons are indicated by asterisks, and N-linked glycosylation sites are indicated by gray shading. Genotype names are indicated to the right of the brackets
Discussion
Previous studies of RSV infection in Thailand used monoclonal antibodies to subtype RSV into A and B subgroups [30, 31]. Recently, there was a molecular and demographic analysis of respiratory syncytial virus infections of inpatients residing in Bangkok, Thailand, conducted during the high season of respiratory tract infection (June – December) in 2007. In our study, we performed a large-scale surveillance study of two groups between 2010 and 2011: an urban population residing in Bangkok and a rural population residing in Chumphae, Khon Kaen Province, in northeastern Thailand. Thus, the purpose of this study was to broaden RSV characterization in Thailand through molecular characterization of a larger and more diverse sample set than has been studied previously. The results showed that the prevalence of RSV infection is greater in Bangkok (27.7 %) than in Khon Kaen (6.8 %), which may be due to the methodology used for sample collection in Khon Kaen. Thus, the throat swab samples from Khon Kaen are not representative of the prevalence of RSV infection. Similar to previous findings [31, 34], our data suggest that the primary targets for RSV infection were children below 2 years of age. Unfortunately, however, the protocol of this study was not designed to test the carrier stage of RSV infection among healthy control subjects.
The epidemiology of RSV in Thailand reveals that this virus predominantly circulates in the rainy season between July and November, with incidence peaks in September and October [30–32, 36]. This epidemiological pattern of RSV spread in Thailand is similar to that of neighboring countries such as Vietnam [37] and Cambodia [17]. Interestingly, in March of 2011, the rate of virus detection was nearly as high as the rate in August despite lying outside the typical period of peak infection. It is believed that this phenomenon was due to an unusually high pressure system originating from China, which resulted in more rainfall and lower temperatures than is usual for March in Thailand [38].
The epidemiological data in the present study demonstrate that RSV-A was the predominant subgroup, and that this subgroup consists of two genotypes: NA1 and ON1 (Fig. 1). This result differs from a report from Thailand in 2007 in which GA2 and GA5 were detected as the predominant RSV-A genotypes [34]. In addition, when we included some of the previous Thailand GA2 samples in our phylogenetic tree, the results showed that these GA2 strains should be re-classified into genotype NA1. Therefore, there may have been no GA2 in 2007. Thus, we reclassified these GA2 strains as NA1 due to the fact that NA1 genotypes were not included in the previous study [34].
Our study has also revealed the prevalence of the new genotype ON1, which is a variant of the NA1 genotype first discovered in Ontario, Canada. This strain has a 72-nucleotide duplication in the second variable region of its G protein, representing the most recently observed modification to the RSV-A subgenotype. A similar duplication phenomenon in the variable region of the G protein was also observed in the 1999 BA genotype of RSV-B [16, 19]. ON1 has so far been detected in five countries from three regions: Canada in North America [16], South Africa in Africa [39] and Japan (nucleotide sequence in the GenBank database), South Korea [40], and now Thailand in Asia. Comparison of the amino acid composition of ON1 in different countries has demonstrated that these strains are completely conserved except for the strain from South Africa, which harbors a lysine (K) at residue 308 (Fig. 3b). To date, the origin and migration of the ON1 strain from North America to Asia and Africa has remained unclear, but further studies on the evolution of this strain in different countries may help to elucidate this question.
The BA genotype was first reported by Trento in 1999, and this strain is characterized by a 60-nucleotide duplication in the second variable region of the G protein [19, 20]. A 2007 report of RSV-B in Thailand revealed that only the BA4 genotype was circulating at that time [34]. In this study, we also detected the BA4 genotype in our dataset, but it was found in only one strain from 2011. However, we also detected the new RSV-B genotypes BA9 and BA10 described by Dapat and colleagues [21]. Furthermore, when we tried to include some of the previous Thailand BA4 strains in our phylogenetic tree, the results indicated that these BA4 strains should be re-classified into genotype BA10. The new BA genotype (BA10) may have begun circulating in Thailand around the same time it emerged in Japan in 2007 [21]. Another interesting discovery was the identification of a new genotype, THB, or Thailand B strain, which is similar to the strains isolated by Deng and colleagues (strain H5598 and H5601) in 2005 [35]. These strains, however, were not officially classified at the time. Our phylogenetic analysis indicates that the strains CU2010/5 and CU2011/81 cluster and are closely related to the H5598 and H5601 strains. This cluster was designated a new genotype according to the bootstrap and p-distance value criteria established by Venter and colleagues [14]. However, further studies, such as whole-genome characterization of the strains, should be performed in order to support and confirm this finding.
In conclusion, our study reports the identification of the RSV-A ON1 genotype and the RSV-B THB genotype in Thailand between 2010 and 2011. This finding indicates the importance of variability in both subgroups, and additional studies on the molecular epidemiology of RSV in Thailand should be continued in order to understand the nature of this virus and the factors that contribute to its virulence. These studies will be of great benefit for the development of vaccines or antiviral agents in the future.
References
Karron RA (2008) Respiratory syncytial virus and parainfluenza virus vaccines. In: Plotkin SA, Orenstein WA, Offit PA (eds) vaccines, 5th edn. Saunders-Elsevier, Philadelphia, pp 1283–1293
Collins PL, Chanock RM, Murphy BR (2001) Respiratory syncytial virus. In: Knip DM, Hoeley P M (ed) Fields virology. Lippincott Williams and Wilkins, Philadelphia, pp 1443–1484
Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehiling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P (2009) The burden of respiratory syncytial virus infection in young children. N Engl J Med 360(6):588–598
Groothuis JR, Hoopes JM, Jessie VG (2011) Prevention of serious respiratory syncytial virus-related illness. I: disease pathogenesis and early attempts at prevention. Adv Ther 28(2):91–109
Hurwitz JL (2011) Respiratory syncytial virus vaccine development. Expert Rev Vaccines 10(10):1415–1433
Levine S, Klaiber-Franco R, Paradiso PR (1987) Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J Gen Virol 68(9):2521–2524
Walsh EE, Hruska J (1983) Monoclonal antibodies to respiratory syncytial virus proteins: identification of the fusion protein. J Virol 47(1):171–177
Mufson MA, Orvell C, Rafner B, Norrby E (1985) Two distinct subtypes of human respiratory syncytial virus. J GenVirol 66(10):2111–2124
Anderson LJ, Hierholzer JC, Tsou C, Hendry RM, Fernie BF, Stone Y, McIntosh K (1985) Antigenic characterization of respiratory syncytial virus strains with monoclonal antibodies. J Infect Dis 151(4):626–633
Cane PA (2001) Molecular epidemiology of respiratory syncytial virus. Rev Med Virol 11(2):103–116
Cristina J, López JA, Albo C, García-Barreno B, García J, Melero JA, Portela A (1990) Analysis of genetic variability in human respiratory syncytial virus by the RNase A mismatch cleavage method: subtype divergence and heterogeneity. Virology 174(1):126–134
Peret TC, Hall CB, Schnabel KC, Golub JA, Anderson LJ (1998) Circulation patterns of genetically distinct group A and B strains of human respiratory syncytial virus in a community. J Gen Virol 79(9):2221–2229
Peret TC, Hall CB, Hammond GW, Piedra PA, Storch GA, Sullender WM, Tsou C, Anderson LJ (2000) Circulation patterns of group A and B human respiratory syncytial virus genotypes in 5 communities in North America. J Infect Dis 181(6):1891–1896
Venter M, Madhi SA, Tiemessen CT, Schoub BD (2001) Genetic diversity and molecular epidemiology of respiratory syncytial virus over four consecutive seasons in South Africa: identification of new subgroup A and B genotypes. J Gen Virol 82(9):2117–2124
Shobugawa Y, Saito R, Sano Y, Zaraket H, Suzuki Y, Kumaki A, Dapat I, Oguma T, Yamaguchi M, Suzuki H (2009) Emerging genotypes of human respiratory syncytial virus subgroup A among patients in Japan. J Clin Microbiol 47(8):2475–2482
Eshaghi A, Duwuri VR, Lai R, Nadarajah JT, Li A, Patel SN, Low DE, Gubbay JB (2012) Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: a novel genotype with a 72 nucleotide G gene duplication. PLoS One 7(3):e32807
Arnott A, Vong S, Mardy S, Chu S, Naughtin M, Sovann L, Buecher C, Beauté J, Rith S, Borand L, Asgari N, Frutos R, Guillard B, Touch S, Deubel V, Buchy P (2011) A study of the genetic variability of human respiratory syncytial virus (HRSV) in Cambodia reveals the existence of a new HRSV group B genotype. J Clin Microbiol 49(10):3504–3513
Blanc A, Delfraro A, Frabasile S, Arbiza J (2005) Genotypes of respiratory syncytial virus group B identified in Uruguay. Arch Virol 150(3):603–609
Trento A, Galiano M, Videla C, Carballal G, García-Barreno B, Merlero JA, Palomo C (2003) Major changes in the G protein of human respiratory syncytial virus isolates introduced by a duplication of 60 nucleotides. J Gen Virol 84(11):3115–3120
Trento A, Viegas M, Galiano M, Videla C, Carballal G, Mistchenko AS, Melero JA (2006) Natural history of human respiratory syncytial virus inferred from phylogenetic analysis of the attachment (G) glycoprotein with a 60-nucleotide duplication. J Virol 80(2):975–984
Dapat IC, Shobugawa Y, Sano Y, Saito R, Sasaki A, Suzuki Y, Kumaki A, Zaraket H, Dapat C, Oguma T, Yamaguchi M, Suzuki H (2010) New genotypes within respiratory syncytial virus group B genotype BA in Niigata. Japan. J Clin Microbiol 48(9):3423–3427
Wertz GW, Collins PL, Huang Y, Gruber C, Levine S, Ball LA (1985) Nucleotide sequence of the G protein gene of human respiratory syncytial virus reveals an unusual type of viral membrane protein. Proc Natl Acad Sci USA 82(12):4075–4079
Johnson PR, Spriggs MK, Olmsted RA, Collins PL (1987) The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: Extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci USA 84(16):5625–5629
Cane PA, Matthew DA, Pringle CR (1991) Identification of variable domains of the attachment (G) protein of subgroup A respiratory syncytial viruses. J Gen virol 72(9):2091–2096
Sullender WM, Mufson MA, Anderson LJ, Wertz GW (1991) Genetic diversity of the attachment protein of subgroup B respiratory syncytial viruses. J Virol 65(10):5425–5434
Cane PA, Pringle CR (1995) Evolution of subgroup A respiratory syncytial virus: evidence for progressive accumulation of amino acid changes in the attachment protein. J Virol 69(5):2918–2925
Melero JA, Garcia-Barreno B, Martinez I, Pringle CR, Cane PA (1997) Antigenic structure, evolution and immunobiology of human respiratory syncytial virus attachment (G) protein. J Gen Virol 78(10):2411–2418
McCullers JA, Saito T (2003) Iverson AR (2004) Multiple genotypes of influenza B virus circulated between 1979 and. J Virol 78(23):12817–12828
Yamashita M, Krystal M, Fitch WM, Palese P (1988) Influenza B virus evolution: co-circulating lineages and comparison of evolutionary pattern with those of influenza A and C viruses. Virology 163(1):112–122
Siritantikorn S, Puthavathana P, Suwanjutha S, Chantarojanasiri T, Sunakorn P, Ratanadilok Na Phuket T, Nawanopparatsakul S, Teeyapaiboonsilpa P, Taveepvoradej S, Pengmesri J, Pongpate S (2002) Acute viral lower respiratory infections in children in a rural community in Thailand. J Med Assoc Thai 85(4):1167–1175
Suwanjutha S, Sunakorn P, Chantarojanasiri T, Siritantikorn S, Nawanoparatkul S, Rattanadilok Na Bhuket T, Teeyapaiboonsilpa P, Preutthipan A, Sareebutr W, Puthavathana P (2002) Respiratory syncytial virus-associated lower respiratory tract infection in under-5-year-old children in a rural community of central Thailand, a population-based study. J Med Assoc Thai 85(4):1111–1119
Fry AM, Chittaganpitch M, Baggett HC, Peret TC, Dare RK, Sawatwong P, Thamthitiwat S, Areerat P, Sanasuttipun W, Fischer J, Maloney SA, Erdman DD, Olsen SJ (2010) The burden of hospitalized lower respiratory tract infection due to respiratory syncytial virus in rural Thailand. PLoS One 5(11):e15098
Samransamruajkit R, Lekhanont P, Sritippayawan S, Prapphal N, Deelodejanawong J, Bhattarakosol P, Poovorawan Y (2003) The occurrence and clinical presentations of RSV subtype in acute bronchiolitis and viral pneumonia among hospitalized infants and young children. 11th Asian congress of Pediatrics
Boonyasuppayakorn S, Kowitdamrong E, Bhattarakosol P (2010) Molecular and demographic analysis of respiratory syncytial virus infection in patients admitted to King Chulalongkorn Memorial Hospital, Thailand, 2007. Influenza Other Respi Viruses 4(5):313–323
Deng J, Zhu R, Quian Y, Zhoa L, Wang F (2006) Sequence analysis of G glycoprotein of human respiratory syncytial virus subtype B strains isolated from children with acute respiratory infections in Beijing, China in years 2000–2004. Chin J Microbiol Immunol 26:1–5. http://europepmc.org/abstract/CBA/606348/reload=0;jsessionid=gYT0lP4lo67Q4yvlXxw4.16. Accessed 16 February 2013 (Abstract)
Olsen SJ, Thamthitiwat S, Chantra S, Chittaganpitch M, Fry AM, Simmerman JM, Baggett HC, Peret TC, Erdman D, Benson R, Talkington D, Thacker L, Tondella ML, Winchell J, Fields B, Nicholson WL, Maloney S, Peruski LF, Ungchusak K, Sawanpanyalert P, Dowell SF (2010) Incidence of respiratory pathogens in persons hospitalized with pneumonia in two provinces in Thailand. Epidemiol Infect 138(12):1811–1822
Yoshida LM, Suzuki M, Yamamoto T, Nguyen HA, Nguyen CD, Nguyen AT, Oishi K, Vu TD, Le TH, Le MQ, Yanai H, Kilgore PE, Dang DA, Ariyoshi K (2010) Viral pathogens associated with acute respiratory infections in central Vietnamese children. Pediatr Infect Dis 29(1):75–77
Climatological Center (2011) Annual weather summary of Thailand in 2011. Meteorological Development Bureau, Thai Meteorological Department. http://www.tmd.go.th/programs%5Cuploads%5CyearlySummary%5CAnnual2011_up.pdf. Accessed 16 February 20
Valley-Omar Z, Muloiwa R, Hu NC, Eley B, Hsiao NY (2013) Novel respiratory syncytial virus subtype ON1 among children, Cape Town. S Afr Emerg Infect Dis. doi:10.3201/eid1904.121465
Lee WJ, Kim YJ, Kim DW, Lee HS, Lee HY, Kim K (2012) Complete genome sequence of human respiratory syncytial virus genotype A with a 72-nucleotide duplication in the attachment protein G gene. J Virol 86(24):13810–13811
Acknowledgments
The study was supported by grants from the Higher Education Research Promotion and National Research University Project of Thailand Office of the Higher Education Commission (HR1155A-55); the National Research Council of Thailand, Center of Excellence in Clinical Virology, Chulalongkorn University; Chulalongkorn University Centenary Academic Development Project, Integrated Innovation Academic Center; Chulalongkorn University Centenary Academic Development Project (CU56-HR01); Ratchadaphiseksomphot Endowment Found of Chulalongkorn University (RES 560530093); Outstanding Professor of the Thailand Research Fund (DPG5480002); and by generous support from the National Research Council of Thailand and King Chulalongkorn Memorial. We would like to thank the Chumphae and Sirindhorn Hospital staff for specimen collection, and Ms. Petra Hirsch for reviewing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Auksornkitti, V., Kamprasert, N., Thongkomplew, S. et al. Molecular characterization of human respiratory syncytial virus, 2010-2011: identification of genotype ON1 and a new subgroup B genotype in Thailand. Arch Virol 159, 499–507 (2014). https://doi.org/10.1007/s00705-013-1773-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1773-9