
Abstract
VPMS1 is a Vibrio parahaemolyticus lytic phage isolated from a marine clam. The 42.3-kb genome was predicted to encode 53 proteins. Comparison of the VPMS1 DNA genome with known phage genomes revealed no similarity; hence, it represents a new VP phage, organized into three differently oriented modules. The module for packaging covers 12 % of the genome, the module for structure covers 31 %, and the module for replication and regulation covers 48 %. The G + C content was 44.67 %. The coding region corresponds to 91 % of the genome, and 9 % apparently does not encode any protein. Thirty genes, constituting 57 % of the genome, had significant similarity to some reported proteins in the protein database; 23 genes, constituting 43 % of the genome, showed no significant homology to any reported protein, and these could be new proteins whose hypothetical functions can be deduced from their position in the genome.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vibrio parahaemolyticus (VP) is a Gram-negative marine bacterium. It is an important food-borne pathogen that can cause illness when improperly prepared seafood is consumed [1]. VP rapidly induces inflammatory gastroenteritis [2], wound infections, and sepsis [3] and can have devastating effects on aquaculture, mainly on shrimp production. Outbreaks in aquaculture have led to high mortality and severe economic loss in all producing countries [4]. Its densities in the environment vary greatly by season and location [5]. Its increasing incidence has become a public-health problem [6]. The use of phages to control undesirable pathogenic bacteria has gained importance in recent years [7]. In this work, we describe the genomic analysis of a highly lytic phage able to control this pathogenic bacterium.
The VP strain corresponds to the 17802 strain in the American Type Culture Collection; phage VPMS1 was obtained from a marine clam (Megapitaria squalida) and showed strong lytic activity in VP cultures [8]. The phage and host were propagated in 2216 culture media, in either broth or agar overlays. The number of VPMS1 particles was determined by the agar double layer method [9]. The phage was concentrated by polyethylene glycol (PEG 8000) precipitation [10] and negatively stained with 2 % (w/v) aqueous uranyl acetate at pH 4.0, on copper grids provided with a carbon-coated Formvar film, and examined by transmission electron microscopy (TEM; EM10, Carl Zeiss) at an accelerating voltage of 80 kV. For DNA analysis, the supernatant was treated with DNAse I (100 U/mL), and RNase A (50 μg/mL), and then with proteinase K (20 mg/mL). Phage DNA was isolated using a modified phenol method [11] and purified using a GENECLEAN Spin Kit (MP Biomedicals, Santa Ana, CA). The identity of VPMS1 DNA was confirmed using EcoRI and Pst I restriction enzymes. DNA quality and quantity was determined on agarose gels and using a photometer (NanoPhotometer Implen, Munich, Germany). To determine the size of the genome of the VPMS1 phage, purified DNA was treated with EcoRI, and the restriction fragments were resolved on agarose gels using lambda phage DNA as a molecular weight reference (BioRad Laboratories, Hercules, CA). The total size of the genomic DNA was determined by adding the sizes of the fragments. DNA was sequenced using a sequencer (Illumina GAIIx at 155x coverage at Base-Clear in Leiden, The Netherlands). The sequence was assembled using “de novo assembly”. Gene sequences were determined with genomics software (CLC Genomics Workbench 4.0, CLC bio, Aarhus, Denmark). When genes overlapped, they were visually inspected, and in some cases removed. Open reading frames (ORFs) were confirmed using the ORF finder from NCBI, and with Sequin software, using the bacterial genetic codes in both cases. Genes were verified using the Heuristic GeneMarkS software [12]. Amino acid sequences were compared to a non-redundant database from NCBI, and search results were visually inspected. Results were taken as significant when e-values were under 0.01. Promoter candidates were determined using the BPROM 0.3.2 software (Softberry, Mount Kisco, NY). A CD search to identify members of protein families was conducted with the NCBI database, and with the Pfam 26.0 software of the Sanger Institute [13]. tRNA scan-SE 1.21 [14], Aragorn v1.2 [15], and tRNA finder (Greengene, University of Massachusetts Lowell) programs were used to search for tRNA genes in the genome.
Results and discussion
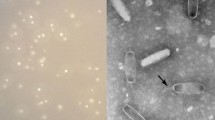
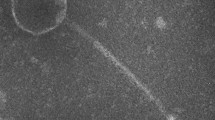
Morphological analysis by TEM showed that VPMS1 had an isometric head (60 nm) and a short tail (10 nm), indicating that it belongs to the family Podoviridae, order Caudovirales (Fig. 1). The nucleotide sequence of VPMS1 has a total size of 42,314 bp (42.3 kb), which is about the average size for Vibrio bacteriophages described on the EMBL-EBI Genome Phage page. The G + C content was 44.67 % – a relatively low value when compared to those found in previously described bacteriophages, but very close to that for VP (45 %) [16]. Comparison of the VPMS1 DNA genome with phage genomes in the NCBI database showed no discernible DNA sequence similarity to any of them; the best match for similarity was about 1 %. Therefore, phage VPMS1 has been designated a new bacteriophage (vibriophage). Analysis of the VPMS1 genome identified 53 putative ORFs; the coding region corresponds to 91 % of the genome and 9 % apparently does not encode any genes. In the genome, 30 genes, constituting 57 % of the genome, showed significant similarity to some proteins reported in the NCBI protein database (Table 1); 23 genes, constituting 43 % of the genome, had no significant homology to reported proteins with known functions (e-values > 0.01). These could be new proteins whose hypothetical functions can be deduced from their position in the genome. Non-repetitive ends (significant) were found at the extremes of the nucleotide sequence, which is important because alignment of repeated regions by NGS is difficult and may lead to errors. In most cases (65 %), coincident genes corresponded to phages infecting Gram-negative bacteria. The presence of tRNAs is important because they facilitate a more rapid overall translation rate; however, they are not found in all phage genomes [17]. For example, the VPMS1 genome does not contain any. Several promoter regions were found and are summarized in Table 1. The VPMS1 genome is organized into three modules, which can be easily identified because some of the genes within them have significant similarity to previously identified genes in the GenBank database (Fig. 2). It is interesting that the orientation of the genes in the VPMS1 genome seems to follow a very specific pattern for grouping. This peculiar gene-oriented organization is not easily seen in phages described in the literature because most of them have indistinct orientations within the modules composing the phage genomes. Previous studies have used the endonuclease gene for phylogenetic analysis [18]; therefore, a phylogenetic analysis was performed using the protein sequence and the genetics analysis software MEGA 5.1. A phylogenetic tree was constructed with the amino acid sequences of endonucleases of some selected phages using neighbor-joining analysis and the maximum composite likelihood model. The results showed a close similarity of VPMS1 to phages infecting enterobacteria, rather than members of the genus Vibrio (Fig. 3). It is worth mentioning that we found a lysogeny-related gene. It is known that temperate phages have a lysogeny module in their genome that contains integrase, repressor, and lysogenic conversion genes [19]. In this case, the lysogeny-related gene was located among the tail fiber genes, which is interesting because we have not seen any reports with a similar lysogeny-related gene organization. However, gene 24 matched the lysogenic conversion protein with low similarity. Also, the integrase gene, which is required for the lysogenic cycle, was not found in the genome. Gene 1 was our candidate for integrase, given that it is located before the helicase and primase genes, but the sequence did not match for integrase. In short, the lysogeny module is not present. The lysogenic conversion protein gene was found, but it is highly improbable that it could be expressed, since there were no integrase or repressor genes. Thus, we conclude that VPMS1 is a lytic phage. The accession number for the complete genome sequence of the VPMS1 phage at NCBI GenBank is JX880072.

Electron micrograph of the Vibrio parahaemolyticus phage VPMS1. The bacteriophage preparation was negatively stained with 2 % uranyl acetate (pH 4.0). Scale bar, 100 nm; Magnification, 50,000×

Genetic and physical organization of the VPMS1 genome. Predicted genes are represented by arrows. Color indicates function (green is the packaging module, brown is the structural module, blue is the replication and regulation module). Promoter regions are indicated by yellow marks. Where known, the functions of genes are indicated (color figure online)

Phylogenetic tree for selected phages constructed from the amino acid sequence of their endonucleases. Vibrio phage VPMS1 seems to be more closely related to enterobacteria phages than to Vibrio phages
References
Matsuda S, Kodama T, Okada N, Okayama K, Honda T, Iida T (2010) Association of Vibrio parahaemolyticus thermostable direct hemolysin with lipid rafts is essential for cytotoxicity but not hemolytic activity. Infect Immun 78:603–610
Sakata J, Kawatsu K, Kawahara R, Kanki M, Iwasaki T, Kumeda Y, Kodama H (2012) Production and characterization of a monoclonal antibody against recombinant thermolabile hemolysin and its application to screen for Vibrio parahaemolyticus contamination in raw seafood. Food Control 23:171–176
Mahoney J, Gerding M, Jones S, Whistler C (2010) Comparison of the pathogenic potentials of environmental and clinical Vibrio parahaemolyticus strains indicates a role for temperature regulation virulence. Appl Environ Microbiol 76:7459–7465
Goarant C, Merien F, Berthe F, Mermoud I, Perolat P (1999) Arbitrarily primed PCR to type Vibrio spp. Pathogenic for shrimp. Appl Environ Microbiol 65:1145–1151
De Paola A, Nordstrom J, Bowers J, Wells J, Cook D (2003) Seasonal abundance of total and pathogenic Vibrio parahaemolyticus in Alabama oysters. Appl Environ Microbiol 69:1521–1526
Ward L, Bej A (2006) Detection of Vibrio parahaemolyticus in shellfish by use of multiplexed real time PCR with TaqMan fluorescent probes. Appl Environ Microbiol 72:2031–2042
Parisien A, Allain B, Zhang J, Mandeville R, Lan C (2007) Novel alternatives to antibiotics: bacteriophages, bacterial cell wall hydrolases, and antimicrobial peptides. J Appl Microbiol 104:1–13
Martínez-Díaz SF, Hipólito-Morales A (2013) Efficacy of phage therapy to prevent mortality during vibriosis of brine shrimp. Aquaculture. doi:10.1016/j.aquaculture.2013.03.007
Kropinski A, Mazzocco A, Waddell T, Lingohr E, Johnson R (2009) Enumeration of bacteriophages by double agar overlay plaque assay. In: Clokie M, Kropinski A (eds) Bacteriophages: methods and protocols. Humana Press, New York, pp 69–76
Carlson K (2005) Working with bacteriophages: common techniques and methodological approaches. In: Kutter E, Sulakvelidze A (eds). Bacteriophages: biology and application. CRC Press, Boca Raton, pp 437–494
Sambrock J, Rusell D (2001) Molecular cloning: a laboratory manual, vol 1. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Besemer J, Borodovsky M (2005) GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucl Acids Res 33:W451–W454
Punta M, Coggill P, Eberhardt R et al (2012) The Pfam protein families. Nucleic Acids Res 40:D290–D301
Schattner P, Brooks AN, Lowe TM (2005) The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucl Acids Res 33:686–689
Laslett D, Canback B (2004) ARAGORN, a program for the detection of transfer RNA and transfer-messenger RNA genes. Nucl Acids Res 32:11–16
Han H, Hinh-Chung W, Kan B, Guo Z, Zeng X, Yin S, Liu X, Yang R, Zhou D (2008) Genome plasticity of Vibrio parahaemolyticus: microevolution of the pandemic group. BMC Genomics. doi:10.1186/1471-2164-9-570
Bailly-Bechet M, Vergassola M, Rocha E (2007) Causes for the intriguing presence of tRNAs. Genome Res 17:1486–1495
Lin Y, Lin C (2012) Genome-wide characterization of Vibrio phage Phi-pp 2 with unique arrangements of the mob-like genes. Bio Med Central Genomics. doi:10.1186/1471-2164-13-224
Brüssow H (2006) Prophage genomics. In: Calendar R, Abedon ST (eds) The Bacteriophages, 2nd edn. Oxford University Press, Oxford, pp 17–25
Acknowledgments
We thank Ira Fogel of Centro de Investigaciones Biológicas del Noroeste, for editorial services, and Diana Barajas for assistance with phage selection, Lina Zermeño for phage production, and Victor Moyrón for technical support. Funding was provided by Consejo Nacional de Ciencia y Tecnología of México (CONACYT grants 52107 and 85033).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramírez-Orozco, M., Serrano-Pinto, V., Ochoa-Álvarez, N. et al. Genome sequence analysis of the Vibrio parahaemolyticus lytic bacteriophage VPMS1. Arch Virol 158, 2409–2413 (2013). https://doi.org/10.1007/s00705-013-1726-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1726-3