
Abstract
Phage Vp_R1 belongs to the family Podoviridae and has a C3 morphotype, with an elongated head with a diameter of 190 ± 1.1 nm and an ultrashort tail with a length of 9 ± 1.2 nm. The double-stranded DNA genome is 112.1 kb long, has a mol% G + C content of 40.3, contains 129 ORFs, and encodes four tRNAs. Phylogenetic analysis suggests that phage Vp_R1 is a novel member of the genus Kuravirus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Bacteriophages with the C3 morphotype are characterized by very long heads that exceed their width by several times. This morphotype is extremely rare among members of the family Podoviridae, and phages of this type constitute only 0.5% of over 5,500 phages that have been examined using electron microscopy [1, 2]. The majority of C3 phages have been characterized by transmission electron microscopy (TEM), but only six of them have been completely sequenced and phylogenetically placed within the genus Kuravirus. This genus typically is found growing on Gram-negative enterobacteria and includes the Escherichia coli phages phiEco32 [23], KBNP1711 (KM044272), ECMP2 (submitted as KBNP135 in GenBank, NC_018859.1), Salmonella phage 7–11, E. coli phage NJ01 and E. coli phage SU10 [13, 18, 19]. phiEco32 is the best-characterized member of this group. The only phage of the C3 type from Gram-positive bacteria is Lactococcus lactis phage KSY1, whose genome has been sequenced and analyzed and shows no similarity to the other C3 phages [6, 24].
Vibrio parahaemolyticus is a zoonotic pathogen that is commonly found in marine animals, seabed sediments, and coastal environments [3]. It can infect numerous types of organisms, including fish, shellfish, and sea cucumbers, and this has caused enormous economic losses in the aquaculture industry [21]. Simultaneously, with the growing popularity of consuming raw or uncooked seafood, V. parahaemolyticus has become the leading source of food poisoning in Asia [5, 11, 16], causing acute gastroenteritis [25]. Lytic bacteriophages have been used as natural agents to kill pathogens and are potentially applicable for use in aquaculture [7, 20]. To date, no genome sequences of C3-like Vibrio phages have been reported. Here, we describe the genomic characteristics of Vibrio phage Vp_R1 and its relationships to phiEco32-like viruses.
The bacterial strain chosen as a host (VP-ABTNL) was isolated from diseased sea cucumbers that were grown in Dalian Bay (Dalian, China) in 2017. Phages were isolated from the same host sample and then propagated in 2216E medium containing the host. Phage genomic DNA was extracted from a high-titer preparation of phage particles (1010 PFU/ml) using the phenol–chloroform isoamyl–alcohol method as described by Sambrook et al. [22]. Phage genomic DNA sequencing was then performed using an Illumina high-throughput sequencing platform (Illumina Hiseq™ 2000, Shenzhen, China).
Morphological observation of phage particles was done using a modification of the protocols described by Goodridge et al. [10]. Purified phage suspensions (≥ 1010 PFU/ml) were adsorbed onto carbon-coated copper grids for 10 min, stained with 0.5% (w/v) uranyl acetate, and then examined using a JEOL-1200EX transmission electron microscope (JEOL USA, Inc., Peabody, MA, USA).
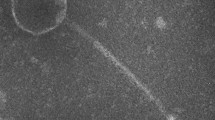
Phage Vp_R1, which was designated as “vB_VpaP_VP-ABTNL-1”, produced clear plaques on a lawn of the host. The plaques had a turbid halo and a diameter of 3.5 mm. Like other C3-like phages, the head exhibited a flattened oval shape. The capsid length of Vp_R1 appeared to be longer than that of phiEco32, 190 ± 1.1 nm, vs. 145 nm, and the tail was a little shorter (9 ± 1.2 nm) (Fig. 1).

Morphology of phage Vp_R1. (A) Plaques of Vp_R1. The scale bar represents 50 mm. (B) Transmission electron micrograph of Vp_R1 negatively stained with 2% w/v uranyl acetate. Black arrows indicate phage tails. The scale bar represents 100 nm
Phage Vp_R1 was found to have a linear double-stranded DNA genome with a length of 112,127 bp (40.32% GC content). Using the GeneMark server (http://topaz.gatech.edu/GeneMark/genemarks.cgi), we identified 129 ORFs, the majority of which (94, 73%) showed no sequence similarity to other viral proteins in the NCBI database. The putative protein functions of the ORFs were annotated by searching against the non-redundant protein database with BLAST-p (http://blast.ncbi.nlm.nih.gov/). Using ARAGORN, Vp_R1 was found to contain four predicted tRNA genes: tRNA-Met (CAT), tRNA–Ile (GAT), tRNA–Arg (CCT), and tRNA-Arg (TCT), ranging from 41,660 to 43,740 bp [15]. A secondary structure visualization was made using forna (http://rna.tbi.univie.ac.at/forna/) (Fig. S1). Phieco32-like viruses all have an arginyl tRNA (AGA), which is considered rare in E. coli [4] (Table 1), and this might allow efficient translation of phage mRNA in the absence of sufficient amounts of the corresponding cellular tRNA could play a subtle role in gene expression [23]. Like in phiEco32, most ORFs of Vp_R1 start with an AUG codon (92.2%), with eight ORFs starting with GUG (6.2%) and two ORFs starting with UUG (1.6%). The three stop codons were present in different proportions, with UAA being the most common (78 ORFs, 60.5%), followed by UAG (28 ORFs, 21.7%), and UGA (23 ORF, 17.8%). In phiEco32, however, the number of ORFs ending with UAG (14 ORFs) is less than the number ending with UGA (32 ORFs).
Of the 129 ORFs, only 35 (27.1%) had functions that could be assigned after sequence alignment with BLASTx in a search against the NR database (Table 2). BLASTn analysis showed a low degree of similarity between Vibrio phage Vp_R1 and other phages, but Vp_R1 showed similarity to predicted genes from the C3-like phages. Furthermore, the genome also comprised four basic segments, including genes coding for structural proteins, DNA metabolism and modification, nucleotide salvage and modification, and other functional genes (Fig. 2). Vp_R1 codes for an assortment of structural proteins, namely portal protein (ORF 91), scaffolding protein (ORF 92), major head protein (ORF 93), tail fiber proteins (ORF 99, ORF 102, ORF 105), phage tail protein (ORF 100), fibritin neck whisker protein (ORF 101), tail tubular protein (ORF 104), and constituent protein (ORF 106). Six proteins were predicted to be related to phage DNA metabolism and modification, including DNA ligase (ORF 16), DNA polymerase (ORF 37, 129), DNA primase/helicase (ORF 38), 5’-3’ exonuclease (ORF 120), and HNH endonuclease (ORF 128). Five proteins were predicted to be related to RNA manipulation, including class Ia ribonucleotide reductase (aerobic) beta subunit (ORF 12), class Ia ribonucleotide reductase (aerobic) alpha subunit (ORF 13), ribose-phosphate pyrophosphokinase (ORF 29), virion-encapsulated RNA polymerase (ORF 113), and RNA polymerase ECF sigma factor (ORF 121). ORF 113 is a large virion-encapsulated RNA polymerase with 6,645 amino acids that is responsible for early gene transcription and may establish the transcription independence of the phage [17]. Interestingly, Vp_R1 has two ribonucleotide reductase proteins, which are likely used by the phage to sustain host metabolism [8]. The Vp_R1 genome encodes an RNA polymerase ECF sigma factor, which has also been found in phage phiEco32 and phage 7-11, and its holoenzyme could recognize promoters of middle or late genes of the virus.

Annotated genome map of Vp_R1. The 129 ORFs are represented as arrows; the direction of each arrow represents the direction of transcription. Proposed modules are based on hypothetical functions predicted from bioinformatic analysis. Green arrows represent the phage structural proteins. Blue arrows represent phage DNA metabolism and modification proteins. Grey arrows represent additional and hypothetical proteins
Vp_R1 codes for at least five proteins related to nucleotide salvage and modification: thymidylate synthase complementing protein, ribose-phosphate pyrophosphokinase, putative polynucleotide 5’ kinase/3’ phosphatase, calcineurin-like phosphoesterase and L-glutamine-D-fructose-6-phosphate aminotransferase-like protein. Notably, ORF 10 encodes a phosphate-starvation-inducible protein (PhoH), which exist in nearly 40% of the marine phage genomes to regulate phosphate uptake and metabolism under low-phosphate conditions, while only 4% of non-marine phage genomes contain this gene, including the C3 phages phiEco32, ECBP2 and SU10 [9]. In addition, holin and lysozyme genes were not found in Vp_R1. Nearly all of the structural proteins are well conserved among C3 phages, especially the scaffolding proteins [19].
Comparisons were made between phage Vp_R1 and other C3-like viruses. Neighbor-joining phylogenetic trees were constructed using MEGA version X [14] based on the amino acid sequences of the phage scaffold protein and major head protein (ORF 93). The trees revealed two major clades, with the phage Vp_R1 sister to KSP100, whose host is Serratia marcescens [12] (Fig. 3). Although Vp_R1 showed a high level of sequence identity to predicted proteins of C3 phages, including terminase large subunit, portal protein, scaffolding protein, major head protein and DNA polymerase, it showed very limited regions where the whole phage DNA sequences were similar, as shown in multiple alignments using the progressive Mauve algorithm (Fig. 4). Interestingly, the tail-related proteins of Vp_R1 had no similarity to those of other C3 phages, and the tail proteins also differed among the other known C3 phages. This suggests that receptor recognition evolves fast in order to allow a wider host range [23].

Neighbor-joining phylogenetic tree based on the scaffolding proteins, showing the relationship between Vp_R1 and other C3 phages. The optimal tree with the sum of branch lengths = 594.07812500 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The analysis included 10 amino acid sequences of C3 phages. All ambiguous positions were removed for each sequence pair. Evolutionary analyses were conducted in MEGA X

Multiple alignments of Vp_R1 and other C3 phages using the progressive Mauve algorithm
Based on morphological and genomic analyses, we propose that phage Vp_R1 should be included as a new C3 phage of the genus Kuravirus.
The GenBank accession number for phage Vp_R1 is MG603697.
References
Ackermann HW (2001) Frequency of morphological phage descriptions in the year 2000. Brief review. Arch Virol 146:843–857
Ackermann HW (2007) 5500 Phages examined in the electron microscope. Arch Virol 152:227–243
Ceccarelli D, Hasan NA, Huq A, Colwell RR (2013) Distribution and dynamics of epidemic and pandemic Vibrio parahaemolyticus virulence factors. Front Cell Infect Mi 3:97
Chen D, Texada DE (2006) Low-usage codons and rare codons of Escherichia coli mini review. Gene Ther Mol Biol 10:1–12
Chen Y, Liu XM, Yan JW, Li XG, Mei LL, Mao QF, Ma Y (2010) Foodborne pathogens in retail oysters in south China. Biomed Environ Sci 23:32–36
Chopin A, Deveau H, Ehrlich SD, Moineau S, Chopin M (2007) KSY1, a lactococcal phage with a T7-like transcription. Virology 365:1–9
Doss J, Culbertson K, Hahn D, Camacho J, Barekzi N (2017) A review of phage therapy against bacterial pathogens of aquatic and terrestrial organisms. Viruses 9:50
Dwivedi B, Xue B, Lundin D, Edwards RA, Breitbart M (2013) A bioinformatic analysis of ribonucleotide reductase genes in phage genomes and metagenomes. BMC Evol Biol 13:33
Goldsmith DB, Crosti G, Dwivedi B, McDaniel LD, Varsani A, Suttle CA, Weinbauer MG, Sandaa R, Breitbart M (2011) Development of phoH as a novel signature gene for assessing marine phage diversity. Appl Environ Microb 77:7730–7739
Goodridge L, Gallaccio A, Griffiths MW (2003) Morphological, host range, and genetic characterization of two coliphages. Appl Environ Microb 69:5364–5371
Harakudo Y, Saito S, Ohtsuka K, Yamasaki S, Yahiro S, Nishio T, Iwade Y, Otomo Y, Konuma H, Tanaka H (2012) Characteristics of a sharp decrease in vibrio parahaemolyticus infections and seafood contamination in Japan. Int J Food Microbiol 157:95–101
Kenshi M, Jumpei U, Shin-Ichiro K, Takako U, Hiroshi H, Shigeyoshi S, Asako M, Hiroshi W, Shigenobu M (2010) Morphological and genetic analysis of three bacteriophages of Serratia marcescens isolated from environmental water. FEMS Microbiol Lett 291:201–208
Kropinski AM, Lingohr EJ, Ackermann H (2011) The genome sequence of enterobacterial phage 7–11, which possesses an unusually elongated head. Arch Virol 156:149–151
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549
Laslett D, Canback B (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32:11–16
Lee JK, Jung DW, Eom SY, Oh SW, Kim Y, Kwak HS, Kim YH (2008) Occurrence of Vibrio parahaemolyticus in oysters from Korean retail outlets. Food Control 19:990–994
Lenneman B, Rothman-Denes L (2015) Structural and biochemical investigation of bacteriophage N4-encoded RNA polymerases. Biomolecules 5:647–667
Li Y, Chen M, Tang F, Yao H, Lu C, Zhang W (2012) Complete genome sequence of the novel lytic avian pathogenic coliphage NJ01. J Virol 86:13874–13875
Mirzaei MK, Eriksson H, Kasuga K, Haggård-Ljungquist E, Nilsson AS (2014) Genomic, proteomic, morphological, and phylogenetic analyses of vB_EcoP_SU10, a podoviridae phage with C3 morphology. PloS One 9:e116294
Nakai T, Park SC (2002) Bacteriophage therapy of infectious diseases in aquaculture. Res Microbiol 153:13–18
Ren H, Li Z, Xu Y, Wang L, Li X (2019) Protective effectiveness of feeding phage cocktails in controlling Vibrio parahaemolyticus infection of sea cucumber Apostichopus japonicus. Aquaculture 503:322–329
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, volume 2. Anal Biochem 186:182–183
Savalia D, Westblade LF, Goel M, Florens L, Kemp P, Akulenko N, Pavlova O, Padovan JC, Chait BT, Washburn MP, Ackermann H, Mushegian A, Gabisonia T, Molineux I, Severinov K (2008) Genomic and proteomic analysis of phiEco32, a novel Escherichia coli bacteriophage. J Mol Biol 377:774–789
Saxelin M, Nurmiaho E, Korhola MP, Sundman V (1979) Partial characterization of a new C3-type capsule-dissolving phage ofStreptococcus cremoris. Can J Microbiol 25:1182–1187
Zhang L, Orth K (2013) Virulence determinants for Vibrio parahaemolyticus infection. Curr Opin Microbiol 16:70–77
Acknowledgements
The authors would like to thank the members of the group of Professor Yigang Tong, Beijing Institute of Microbiology and Epidemiology, for help with sequence analysis.
Funding
This work was financially supported by the National Public Science and Technology Research Funds Projects of Ocean (Grant No. 201405003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals by any author.
Additional information
Handling Editor: T. K. Frey.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Fig. S1 Secondary structure visualization of tRNAs of Vp_R1. A. tRNA-Met (CAT). B. tRNA–Ile (GAT). C. tRNA- Arg (CCT). D. tRNA-Arg (TCT).
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ren, H., Li, Z., Xu, L. et al. Genome sequence analysis of Vibrio parahaemolyticus lytic phage Vp_R1 with a C3 morphotype. Arch Virol 164, 2865–2871 (2019). https://doi.org/10.1007/s00705-019-04364-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-019-04364-0