
Abstract
This is the first report of the genetic diversity within ilarvirus subgroup 1 from eastern Australia. It supports the separation of tobacco streak virus (TSV) strains from parthenium (Parthenium hysterophorus) and crownbeard (Verbescina encelioides) based on serology and host specificity. It has confirmed one previously described strain of TSV as a member of the species Strawberry necrotic shock virus and another as a new subgroup 1 ilarvirus, ageratum latent virus (AgLV), from Ageratum houstonianum. A multiplex RT-PCR showed that the genetically distinct strains of TSV and AgLV were commonly found in symptomless infections in virus-specific alternative weed hosts growing over a wide geographical range in eastern Australia. TSV has been one of the most damaging viruses in Australian oilseed and pulse crops in recent years, and this study has provided the taxonomic knowledge essential for the development of control programs for these viruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ilarviruses (family Bromoviridae) have a positive-sense, single-stranded RNA genome consisting of three linear segments (RNA-1 to -3). The 1a (viral replicase) protein is encoded by RNA-1, and the 2a (RNA-dependent RNA polymerase, RdRp) and 2b proteins are encoded by RNA-2. The 3a (cell-to-cell movement) protein (MP) and 3b coat protein (CP) are encoded by RNA-3, although the CP is translated from the sub-genomic RNA-4, which is derived from RNA-3 [1].
The demarcation criteria for ilarvirus species combine serological relatedness, host range and genome sequence similarity [1], although there is currently no specified level of sequence similarity to distinguish species, strains or subgroups. Ilarvirus species have been assigned to subgroups based on serological relatedness, and ilarvirus strains belonging to the same species have also been reported based on serological differences, host specificity or geographical location. Members of subgroups 1 and 2 have a unique second open reading frame (ORF) on RNA-2 for the putative 2b protein, which is absent from other ilarvirus subgroups. Recent phylogenetic studies on ilarviruses have suggested greater emphasis on sequence identity in RNA-3 coding regions [2], the RNA-1 and RNA-2 sequences [3] or whole-proteome (all putative proteins) analysis [4] for defining species.
Tobacco streak virus (TSV), the type member of the genus Ilarvirus, was identified in 1936 [5] and was the only described member of subgroup 1 until the mid 1990s, when the development of genome sequencing techniques resulted in the creation of several additional species. The type isolate of TSV was originally isolated from white clover (TSV-WC; [6]). Species within subgroup 1 include Tobacco streak virus, Strawberry necrotic shock virus (SNSV), Parietaria mottle virus (PMoV), Blackberry chlorotic ringspot virus (BCRV) [7] and the proposed species “Bacopa chlorosis virus” [8]. Two subgroup 1 viruses have been reported previously from Australia: TSV [9] and, more recently, SNSV [10].
In Australia, TSV was first described in the early 1970s as the cause of a sporadically occurring disease of commercial tobacco crops in south-eastern Queensland [9]. Three distinct strains of TSV have been described from Australia and were shown to differ in relation to serology, host range and thrips species transmission efficiency [9, 11–13]. The most commonly reported strain was originally referred to as the Asclepias strain (TSV-As; [13]) and later as the Ageratum strain (TSV-Ag; [12]) due to its high incidence in Ageratum houstonianum (ageratum) near TSV-affected tobacco crops [14]. A strain originally isolated from strawberry, TSV-S [11, 13], has recently been demonstrated to actually be SNSV [10]. A third strain, TSV-A, was isolated from Ajuga reptans in Melbourne, Victoria, in the late 1970s and was shown to be serologically distinct from strain TSV-Ag [15]. However, no further reports of this TSV-A strain have been made, and it is unclear if it is still present in Australia. Similarly, TSV-S (SNSV) has recently been reported from reference collections but has not been reported in natural infections since the late 1970s [10].
We recently reported a TSV strain from central Queensland causing significant economic losses in sunflower and mung bean crops over several seasons [16]. This TSV strain (hereafter referred to as TSV-parthenium) was commonly found in asymptomatic Parthenium hysterophorus, and this prolific weed is considered to be a major source of inoculum for disease epidemics in surrounding crops [17]. A partial RNA-3 sequence of the TSV-parthenium strain indicated that it was most closely related to a soybean TSV isolate from Brazil [18], but preliminary results demonstrated that TSV-parthenium was genetically divergent from TSV-Ag from south-east Queensland. In the course of the present work, we found a further distinct TSV strain commonly infecting Verbescina encelioides (crownbeard), hereafter referred to as TSV-crownbeard. These results suggested that there was significant diversity in TSV strains present in eastern Australia. With the recent discovery of the TSV-parthenium and TSV-crownbeard strains, there were a total of five distinct ilarvirus subgroup 1 members, which could constitute TSV strains or members of other ilarvirus species. A partial RNA-3 sequence was published for only SNSV (TSV-S) [10] and TSV-parthenium [17].
The main focus of this paper is to describe the genetic diversity of subgroup 1 ilarviruses from eastern Australia and in doing so clarify the taxonomy of a range of strains previously reported as TSV. Serological and host data are used to support conclusions made from the phylogenetic analyses.
Materials and methods
Virus isolates
Leaf material was collected between 2006 and 2010 from many locations in central and south-east Queensland and northern New South Wales (Table 1). Samples were either collected from locations isolated from cultivated areas or close to crops affected by TSV. The isolates -1973 from parthenium, -1998 from ageratum, and -2334 from crownbeard were selected for further detailed investigation as typical representatives of distinct ilarvirus strains from the three commonly found weed hosts. They were propagated and maintained in N. tabacum cv. Xanthi plants in a glasshouse for use as diagnostic controls and for additional characterization experiments. All isolate numbers refer to samples lyophilised and stored at −20 °C in the Queensland Department of Agriculture, Fisheries and Forestry plant virus collection.
ELISA
Fresh or lyophilised samples were tested for TSV by ELISA (AGDIA ELISA reagent set, catalogue no. SRA25500/0500) as per the manufacturer’s instructions with minor modifications. Coating antibodies and conjugate were used at a dilution of 1:500, and conjugate was cross-absorbed with a mix of healthy Helianthus annuus and Nicotiana tabacum cv. Xanthi, each diluted 0.1 g per 10 ml of PBS-T. Leaf tissue was extracted at 0.1 g per 1 ml of PBS-T containing 2 % polyvinylpyrrolidone, with reaction volumes of 50 or 100 μl in duplicate. Absorbance values (A410 nm) were measured using a Thermo Electron Corporation Multiskan EX plate reader, and values greater than three times the mean of the healthy controls were considered positive.
Polymerase chain reaction (PCR)
To design PCR primers (Table 2), previously published RNA-1, -2, and -3 sequences for TSV-WC, SNSV-MD and PMoV (Table 3) were aligned using the MUSCLE algorithm [19], and conserved regions were selected by eye. Various combinations of primer pairs were used to amplify overlapping fragments from the four reference isolates characterised (see below).
Total nucleic acid extracts (TNAEs) were prepared from lyophilised leaf tissue using a BioSprint 15 workstation (QIAGEN, catalogue no. 9000850) with a BioSprint 15 Plant DNA kit (catalogue no. 941514) as per the manufacturer’s instructions but without the use of RNase A in the RLT buffer. SuperScript III reverse transcriptase (Invitrogen) was used to prepare cDNA as per the manufacturer’s instructions. PCR was done using 10 pmoles of each primer, 1 unit native Taq DNA polymerase (Invitrogen), 1.75 mM MgCl2, 200 mM dNTPs and 2 μl of cDNA template in a 25-μl reaction volume. Due to the large number of combinations of different primer pairs and templates, generic ramped annealing temperature cycling parameters [20] were used for as many primer pairs as possible, consisting of an initial denaturation of 95 °C for 60 s, then 35 cycles of 95 °C for 20 s, 62 °C for 20 s, 56 °C for 10 s and 72 °C for 45 s, followed by a final extension of 72 °C for 3 min on a C1000 Thermal Cycler (Bio-Rad).
Reference isolates selected for characterisation of the whole genome
Although isolate TSV-1973 was originally collected from sunflower displaying typical TSV symptoms (Table 1), a preliminary partial RNA-3 sequence demonstrated that it resembled a strain commonly found in parthenium [17], and as such, TSV-1973 was retained as a reference isolate for the TSV-parthenium strain. Isolates -2334 (TSV-crownbeard) and -840 (SNSV; TSV-S) were selected for characterisation. Isolate -1998 was selected as the reference isolate for the proposed new subgroup 1 ilarvirus species, “Ageratum latent virus” (AgLV), previously reported as strain TSV-Ag.
Multiplex RT-PCR for segment RNA-3
To differentiate TSV-parthenium, TSV-crownbeard and AgLV and to enable identification of mixed infections, a multiplex RT-PCR was developed. A degenerate reverse primer (TSVRNA3.1982R) was used for cDNA synthesis for all strains and then combined with strain-specific forward primers for PCR. To test if the multiplex RT-PCR could detect mixed infections of these ilarvirus strains, artificial mixes were made by combining TNAEs in roughly equal proportions prior to cDNA synthesis. PCR mix and temperature cycling were as described above, but with the following optimised mix of primers: 200 nM TSVRNA3.1982R, 300 nM CQTSVF, 100 nM SEQTSVF and 80 nM CrbTSVF (Table 2).
5′- and 3′-terminal sequencing
The 5′ and 3′ termini of each RNA segment for each of the reference isolates, -1973, -1998, -2334 and -840 were determined utilising a single oligo, Adaptor2 (5′-PO4-CGACATAACCTTGCACATGACTCGAACGGAAATCCCATATATTGCCGAGACAG-3′) with a 3′ C-3 spacer to stop self-ligation. Adaptor2 was either ligated directly to the 3′ terminus of RNA molecules, including viral genomic RNA, or to the 3′ end of cDNA made to the 5′ end of genomic RNA. Ligation reactions were modified from previously described methods [21, 22].
For the 5′ terminus, cDNA was prepared using primers binding about 500 nt from the predicted 5′ terminus and with 7 μl of TNAE. To remove excess ssRNA and RNA from the RNA/cDNA duplex, 1 U of RNase A (QIAGEN) and 2 U of RNase H (Epicentre) were added to the cDNA reaction for 30 min at 37 °C, followed by 10 min at 65 °C to inactivate the enzymes. To remove unincorporated primer, the reaction was cleaned using a PCR purification kit (QIAGEN) as per the manufacturer’s instructions and eluted in 30 μl of 10 mM Tris-Cl pH 8.5. Ligations were done at 24 °C for 18 h in 1 × T4 DNA ligase buffer containing a final concentration of 1 mM ATP (New England Biolabs; catalogue no. B0202S), 25 % (wt/vol) PEG 8000, 10 μg of bovine serum albumin per ml, 10 pmoles of Adaptor2 oligo, 10 U T4 RNA ligase (Epicentre), 1 mM hexammine cobalt(III) chloride (Sigma-Aldrich) and made up to a reaction volume of 35 μl with cleaned cDNA. Ligations were terminated at 65 °C for 10 min. The preparation was then used in PCR with a primer complementary to the Adaptor2 oligo, AdaptR1 (Table 2), and a virus-specific downstream primer, preferably internal to the primer used for cDNA synthesis. Nested PCR using a second virus-specific primer and AdaptR2 was sometimes required to obtain a distinct PCR band.
For the 3′ terminus, ligations were carried out as described above but with 20 pmoles of Adaptor2 oligo, 15 U T4 RNA ligase and reaction made up to volume with TNAE. Following termination of ligation unincorporated Adaptor2 oligo and enzyme were removed by passage through a Sephadex G15 column with elutions made in H2O and 1 μl was then used in cDNA synthesis reactions with primer AdaptR1. The resulting cDNA was used in PCR as described above.
In the case of RNA-3 for SNSV-840, the above method did not work for obtaining the 3′ terminal sequence, and polyadenylation of the 3′ end was carried out using E. coli poly(A) polymerase (Ambion, Cat. No. AM1350) as per the manufacturer’s instructions, followed by cDNA synthesis with primer Poty 1 [23].
Sequencing and analysis
PCR products were analysed by electrophoresis on 0.5× TBE agarose or using an E-Gel® CloneWell™ 0.8 % SYBR Safe™ gel on an E-Gel® iBase™ Power System (Invitrogen, catalogue no. G6618-08 and G6400) as per the manufacturers’ instructions and extracted in water. PCR products were sequenced directly using an Applied Biosystems Inc. automated sequencing system (Australian Genome Research Facility, Brisbane) to provide at least twofold coverage of the target regions.
A representative selection of complete genome sequences of currently recognised ilarvirus subgroup 1 members [7] including TSV, SNSV, BCRV and PMoV, along with isolates characterised in the present work, were used for phylogenetic analysis (Table 3). Nucleotide and putative amino acid sequences were aligned using the MUSCLE algorithm [19] included in the MEGA5 software package [24]. MEGA5 was also used to calculate pairwise uncorrected genetic distances between sequences in the alignments. For nucleotide sequence alignments, putative ORFs were translated into proteins, aligned by MUSCLE and returned to the original nucleotide sequence. Non-coding regions were aligned by MUSCLE and the resulting nucleotide alignments were used for phylogenetic analysis. Spinach latent virus (SpLV) and Prunus necrotic ringspot virus (PNRSV) were included as out groups from ilarvirus subgroups 2 and 3, respectively (Table 3).
Phylogenetic relationships were inferred using the maximum-likelihood method as implemented in RaxML [25] and PhyML 3.0 [26]. GTR-GAMMA was specified as the model of evolution in both programs. The RaxML analyses were run with a rapid bootstrap analysis using a random starting tree and 1000 maximum-likelihood bootstrap replicates. PhyML analysis was implemented using the ATGC bioinformatics platform (available at: http://www.atgc-montpellier.fr/phyml/), with SPR and NNI tree improvement and support obtained from an approximate likelihood ratio test [27]. For analysis of the nucleotide sequence of the complete genome, the three complete RNA segments were included as separate partitions in a maximum-likelihood analysis so each locus could be run under different optimal model parameters. A similar method was used for protein analysis (whole proteome) with the five putative proteins (loci) included as separate partitions in a maximum-likelihood analysis. The resulting trees were observed with FigTree (available at http://www.tree.bio.ed.ac.uk/software/figtree/). Nucleotide sequence identity searches of the GenBank database were done using the Basic Local Alignment Search Tool (BLAST; [28]). Recombination analysis was carried out using the program RDP3 with the default parameters [29].
Results
Field surveys and identification by ELISA and multiplex RT-PCR
All isolates listed in Table 1 were positive by TSV ELISA and all ELISA-positive samples of P. hysterophorus, A. houstonianum and V. encelioides were symptomless. When the reference isolates -1973 (TSV-parthenium), -2334 (TSV-crownbeard) and -1998 (AgLV) were maintained in tobacco, consistent differences in relative A410nm values were observed in TSV ELISA, suggesting serological variability. From three independent tests, A410nm values were 17 to 21 times the average of the healthy controls for isolate -1998 (AgLV), 329 to 726 times for isolate -1973 (TSV-parthenium) and 740 to 1113 times for isolate -2334 (TSV-crownbeard). The relatively low A410nm values for isolate -1998 were similar to that of SNSV-840, which was nine times the average healthy control in a single test.
Using the multiplex RT-PCR, products of unique size were amplified (Fig. 1a) from the reference samples of TSV-parthenium (isolate -1973, 921 bp), AgLV (isolate -1998, 743 bp) and TSV-crownbeard (isolate -2334, 571 bp). The three strains were found in many samples across a wide geographical area, each spanning several hundred kilometres from north to south, apparently coinciding with, and possibly endemic throughout, the geographical range of their respective alternative host (Table 1). The TSV-parthenium strain was never found in crownbeard, nor vice versa, even though both strains and hosts were found in central Queensland, often occurring in the same locations. The distribution of AgLV had no overlap with the TSV strains and was found in ageratum, which occurs in higher-rainfall areas, mostly east of the Great Dividing Range along the eastern seaboard of Australia. While no natural mixed infections of the three strains were found, all were detected by multiplex RT-PCR in artificial mixes, although there was a moderate decrease in amplification of AgLV when all three strains were combined (Fig. 1b). A summary of the multiplex RT-PCR results is shown in Table 1, and gel electrophoresis results for a selection of the parthenium, ageratum and crownbeard samples are shown in Fig. 1a.

a Multiplex RT-PCR for segment RNA-3 of TSV-parthenium, TSV-crownbeard and AgLV. Lanes 1 to 6 are TSV-parthenium isolates -1973, -2084, -2087, -2103, -2105 and healthy parthenium. Lanes 7 to 11 are AgLV isolates -1998, -2291, -2403, -2558 and healthy ageratum. Lanes 12 to 17 are TSV-crownbeard isolates -2334, -2282, -2338, -2400, healthy crownbeard and water control. Marker lanes are 100-bp ladder (Fermentas). b Multiplex RT-PCR for segment RNA-3 of mixed TSV-parthenium (isolate -1973), TSV-crownbeard (isolate -2334) and AgLV-1998. Lane 1, TSV-parthenium and AgLV; lane 2, AgLV and TSV-crownbeard; lane 3, TSV-parthenium and -crownbeard; lane 4, TSV-parthenium and -crownbeard and AgLV; and lane 5, water control. Marker lanes are 100-bp ladder (Fermentas)
Characterisation of complete genomes of representative isolates
The PCR primers (Table 2) for RNA-1, -2 and -3 worked for all four reference isolates characterised: isolate -1973, -1998, -2334 and -840. The use of a single oligonucleotide, Adaptor2, to attach to both 5′ and 3′ ends was an effective, low-cost means for determining the terminal sequences as described. The option of A-tailing the 3′ end by polyadenylation was also effective for templates that were problematic with the first method such as the 3′ end of RNA-3 for isolate -840. The complete nucleotide (nt) sequences of RNA-1, -2 and -3 were determined for isolates -1973, -1998, -2334 and -840, and partial RNA-1, -2 and -3 sequences were determined for isolate -1025. Partial RNA-3 sequences were also determined for archived isolates -837 and -835, with GenBank accession numbers shown in Table 3. For the four isolates for which complete genomes were determined, a summary of genome features and a comparison of their amino acid sequences to that of the type ilarvirus TSV-WC are shown in Table 4.
RNA-1 of the four completely sequenced isolates contained the putative ORF for the 1a (replicase) protein, within which the methyl transferase-like and helicase-like signatures were found [30, 31]. RNA-2 encoded the putative 2a (RdRp) protein, with the conserved polymerase signature [30], including the region ASGDDSLI, which is highly conserved among ilarviruses. RNA-2 also encoded the putative 2b protein. RNA-3 contained the putative ORFs for the 3a MP and 3b CP. The conserved folded stem-loop structures and AUGC-like motifs at the 3′ end of each RNA segment [32] were identified for the characterised isolates. Using the motif numbering proposed by Bol [32], motif 2 was highly conserved as AUGC for every RNA segment characterised, while motif 1 was found to be AUGC or AAGC. Motif 3 was most variable, but with conserved U and C at the third and first nucleotide as (A/G/U)U(A/G/U)C. Isolate -1998 was unique in having UUUC for motif 3 of each RNA segment.
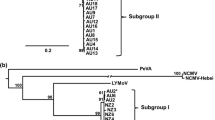
Phylogenetic analysis
A phylogram produced using the maximum-likelihood analysis with RaxML for the complete nt genome sequence of the newly characterised TSV isolates -973 and -2334, AgLV-1998 and SNSV-840, and other ilarviruses showed a number of strong relationships (Fig. 2). The topology of this phylogram was strongly supported by maximum-likelihood analysis with RaxML for the complete proteome sequence of the same samples, and also by maximum-likelihood analysis with PhyML for the separate complete RNA-1 and RNA-2 nt sequences. The recognised subgroup 1 members formed a well-supported clade that was separate from the subgroup 2 and 3 out group representatives, SpLV and PNRSV. AgLV-1998 was sister to PMoV within the subgroup 1 clade but had only 69-71 % total genome nt sequence identity to PMoV, SNSV or TSV-WC. By comparison, the recognised subgroup 1 members BCRV and SNSV shared 81 % total genome nt identity. The phylogram produced for the complete RNA-3 nt sequence using maximum-likelihood analysis with PhyML showed a slightly different topology, with the BCRV/SNSV clade grouping with the AgLV/PMoV clade instead of the TSV clade (data not shown).

Phylogram obtained from a maximum-likelihood analysis in RAxML for the complete genome nucleotide sequence of the ilarviruses characterised in this study and other previously published ilarviruses. The complete sequence of each RNA segment was included as separate partitions in a maximum-likelihood analysis. The scale bar represents the number of nucleotide substitutions per site. Maximum-likelihood support values (> 50 %) from RAxML 1000 bootstrap replicates are shown above the nodes as the upper values. The lower values (above the nodes) are from the RAxML analysis of the separate partitioned putative protein sequences (whole proteome) for which the phylogram topology was the same as that shown. There is no corresponding scale bar for the protein analysis. The same phylogram topology was obtained from maximum-likelihood analyses in PhyML for the complete RNA-1 and RNA-2 nucleotide sequences. Maximum-likelihood support values > 50 % from PhyML are shown below the nodes for RNA-1 (closest to the node) and RNA-2. SpLV and PNRSV were used as out-groups to root the phylograms. GenBank accession numbers are listed in Table 3 and virus acronyms are defined in body of the text
Of the complete genome sequences presented in this study, TSV-2334 (TSV-crownbeard) was the most closely related to the type member TSV-WC (Fig. 2), with a total-genome nt sequence identity of 88 %. TSV-2334 had 81 % total-genome nt sequence identity to TSV-1973. A partial RNA-3 sequence for isolate -835 had 98 % nt sequence identity to TSV-2334 over a 1141-nt overlap, demonstrating that the TSV-crownbeard strain has been present in Australia since at least 1975, when isolate -835 was collected.
TSV-1973 (TSV-parthenium) along with isolates -1974 and -2012 from related studies [16, 17] and a Brazilian isolate, TSV-BR [18], share very high identity (99-100 %) in the partial RNA-3 sequence over a 762-bp overlap, and all appear to be isolates of a genetically divergent strain of TSV. The complete RNA-1, -2 and -3 sequences of TSV-1973 had relatively low nt sequence identity (closest matches 77 %- 81 %) to all other published complete RNA-3 TSV sequences (data not shown), including the type isolate TSV-WC. All phylogenetic analyses (Fig. 2) and putative aa identities (Table 4) indicated that TSV-1973 is distinct from the type member TSV-WC (Fig. 2).
The six separate phylogenetic analyses demonstrated that AgLV-1998 should be considered a member of a distinct species in ilarvirus subgroup 1. BLASTN searches for AgLV-1998 showed 76 % nt identity to BCRV for RNA-1 and RNA-2, and 72 % to TSV-WC for RNA-3. The partial RNA-3 sequence of isolate -837 had 99 % nt sequence identity to AgLV-1998 over a 584-bp overlap, indicating that isolate -837 should be considered an isolate of AgLV. The closest aa identities for each of the putative proteins of AgLV-1998 were 81 % (replicase, PMoV), 76 % (RdRp, PMoV), 68 % (2b, BCRV), 79 % (MP, TSV-WC), and 75 % (CP, TSV-WC).
Isolate -840, a reference sample referred to as strain TSV-S [13], was shown to be an isolate of SNSV in all phylogenetic analyses, supporting its previous identification based on partial RNA-3 sequence data [10]. Isolate SNSV-MD from Maryland, USA, is the only other completely sequenced isolate of SNSV, with which SNSV-840 shares 96 %, 95 % and 93 % nt sequence identity for the complete RNA-1, -2 and -3, respectively. However, SNSV-840 was more closely related to a Mississippi, USA, SNSV isolate (AY363233), with 99 % nt sequence identity for the partial RNA-3 sequence [10] over a 669-bp overlap.
Isolate -1025, a reference sample from Ajuga reptans referred to as TSV-A [15], grouped most closely with the type member TSV-WC in the putative 3b CP ORF, with 99 % aa identity. However the putative 3a MP of isolate -1025 shared only 85 % aa sequence identity with TSV-WC. Partial RNA-1 and RNA-2 nt sequences of isolate -1025 were also divergent from TSV-WC, with 90 % and 87 % identity over 642-bp and 1100-bp overlaps, respectively. Interestingly, a partial RNA-2 sequence for a rhubarb TSV isolate from the USA (HQ130450) had 99 % nt sequence identity with isolate -1025 over a 419-bp overlap, but the relationship between these two isolates is unclear for other regions of the genome. For isolate -1025, a likely recombination event on RNA-3 of an unknown major parent with TSV-WC as minor parent was strongly supported by the RDP [29], GENECONV [33], BootScan [34], MAXCHI [35] and SiScan [36] methods, all with p-values less than 1 × 10−12, and also by the CHIMAERA [37] method, with a p-value of 6 × 10−5. No other putative recombination events were strongly supported for any of the other strains of TSV, AgLV or SNSV in this study.
Partial RNA-1, -2 and -3 sequences have been reported for an isolate of TSV from South Africa [38], but these were not available on the GenBank database. No close relationship was found between the South African TSV isolate and the characterised isolates presented here. For example, for a 691-bp overlap of the partial RNA-3, the South African TSV isolate shared 96 %, 89 % and 80 % nt sequence identity with TSV-WC, isolate -2334 and isolate -1973 respectively.
Discussion
This study has clarified the genetic identity of several ilarvirus isolates from eastern Australia, some of which had previously been identified as strains of TSV. In the process, one of these strains (TSV-Ag) was shown to be a member of a new ilarvirus species, tentatively named “Ageratum latent virus”, while another (TSV-S) was shown to be a member of the species Strawberry necrotic shock virus. Two other strains of TSV were also described from central Queensland, TSV-parthenium and TSV-crownbeard. TSV-parthenium, TSV-crownbeard and AgLV display serological differences and are common and widespread in eastern Australia in symptomless weed species, which appear to be specific for each. This new knowledge of the taxonomy and biology of these strains is an essential prerequisite for the development of control programs for these viruses. These results also underline the value of retaining reference and voucher specimens, as previously published biological data could be linked to genetic analysis through these specimens.
The complete genomes of the reference isolates -1973 (TSV-parthenium), -2334 (TSV-crownbeard), AgLV-1998 and SNSV-840, and the partial genome of isolate -1025 (TSV-A) revealed that significant genetic diversity exists in eastern Australia and adds greatly to the known diversity of ilarvirus subgroup 1 members. The degree of genetic diversity found in this study supports the proposition by Tzanetakis et al. [39] that the cluster of TSV-like strains may represent several distinct species.
Sequence data demonstrated that isolate -1998 from ageratum is closely related to reference isolate -837, which was also referred to as the Ageratum strain (TSV-Ag) by Klose et al. [12], and both are members of a new subgroup-1 ilarvirus species. With consideration of its previous naming as Ageratum TSV, along with our observations and the previous reports of this strain being commonly found as symptomless infections in Ageratum houstonianum [12, 14] we propose the name “Ageratum latent virus” (AgLV). The phylogenetic results of this study and previous work showing serological and host-range differences between AgLV and other TSV strains [13], along with the apparent geographical isolation of AgLV from the TSV-parthenium and -crownbeard strains also support the creation of this new species and satisfy the requirements for the demarcation of new ilarvirus species [1].
The differential reactions observed for the TSV strains, AgLV and SNSV in TSV ELISA correlate with the relative identities of their respective putative coat proteins to that of TSV-WC but complicate definitive diagnosis. The use of the multiplex RT-PCR eliminates this ambiguity in diagnosis and is being used in a related investigation to assist with screening of sunflower germplasm for tolerance to the TSV-parthenium strain.
The TSV-parthenium and TSV-crownbeard strains and AgLV were commonly found across wide geographical ranges in strain-specific alternative weed hosts in symptomless infections. This was also the case for many more field samples of parthenium that we tested from central Queensland [17]. These virus strains have no apparent impact on their respective strain-specific weed hosts, suggesting a stable interaction. The presence of the viruses is only apparent when they induce disease symptoms in nearby susceptible plants of other species, including crops. The possible reasons why TSV-parthenium has not been found in natural infections of crownbeard or TSV-crownbeard in parthenium are not well understood. Both plant host species, with their respective TSV strains, often grow in direct contact in the same locations throughout large areas of central Queensland, and the same common thrips vector species have been found on both hosts (data not shown).
The use of alternative host names, parthenium and crownbeard, to describe the TSV strains from this study has been adopted, as it appears that these are the major hosts for these TSV strains over the geographical distribution of these hosts in Australia. However, given the diversity of TSV strains now published, care needs to be taken in how these are referred to. For example, parthenium in India is also considered to be the principle source of TSV for the development of disease epidemics in nearby crops [40], but the TSV strains reported from India are genetically distinct from the TSV-parthenium strain reported here from Australia.
The current wide geographical distribution of TSV-crownbeard in V. encelioides in central Queensland and the identification of a closely related isolate collected in 1975 from coastal north Queensland would indicate that the TSV-crownbeard strain has been present in this region for several decades. However, severe TSV disease epidemics in sunflower and mungbeans have only been reported since the early 2000s, and these epidemics have only been associated with the TSV-parthenium strain [16], so it seems unlikely that the TSV-crownbeard strain has caused severe disease epidemics in the past.
While substantial previous work has been done to characterise the biology of AgLV, further investigation of the TSV-parthenium and -crownbeard strains are being conducted to further characterise their natural host range, biological characteristics including seed and thrips transmission, and their respective roles in the development of disease epidemics in central Queensland crops. We are also conducting further studies to investigate possible mechanisms that may be restricting these two strains to their respective weed hosts in nature and to determine whether reassortments of RNA segments can be found in natural infections or induced by artificial mixed infections.
References
King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (2011) Virus taxonomy: ninth report of the international committee on taxonomy of viruses. Elsevier Academic Press, San Diego
Codoñer FM, Cuevas JM, Sánchez-Navarro JA, Pallás V, Elena SF (2005) Molecular evolution of the plant virus family Bromoviridae based on RNA3-encoded proteins. J Mol Evol 61(5):697–705
Boulila M (2009) Recombination structure and genetic relatedness among members of the family Bromoviridae based on their RNAs 1 and 2 sequence analyses. Virus Genes 38(3):435–444
Codoñer FM, Elena SF (2006) Evolutionary relationships among members of the Bromoviridae deduced from whole proteome analysis. Arch Virol 151(2):299–307
Johnson J (1936) Tobacco streak, a virus disease. Phytopathology 26:285–292
Fulton RW (1967) Purification and some properties of Tobacco streak and Tulare apple mosaic viruses. Virology 32:153–162
ICTV (2011) ICTV Master List 2011. http://talk.ictvonline.org/files/ictv_documents/m/msl/4090.aspx. Accessed 2 July 2012
Maroon-Lango CJ, Aebig J, Hammond J, Hsu HT (2006) Molecular and biological characterization of a novel ilarvirus in bacopa. Phytopathology 96(6):S73
Greber RS (1971) Some characteristics of tobacco streak virus isolates from Queensland. Qld J Agri Anim Sci 28:105–114
Sharman M, Constable F, Perera R, Thomas J (2011) First report of Strawberry necrotic shock virus infecting strawberry (Fragaria vesca) from Australia. Australas Plant Dis Notes 6:54–56. doi:10.1007/s13314-011-0018-6
Greber RS (1979) Virus diseases of Queensland strawberries and the epidemiological effects of the strawberry runner approval scheme. Qld J Agric Anim Sci 36(1):93–103
Klose MJ, Sdoodee R, Teakle DS, Milne JR, Greber RS, Walter GH (1996) Transmission of three strains of Tobacco streak ilarvirus by different thrips species using virus-infected pollen. J Phytopathol 144:281–284
Sdoodee R (1989) Biological and physicochemical properties of Tobacco streak virus. PhD thesis, University of Queensland, Brisbane
Greber RS, Klose MJ, Teakle DS, Milne JR (1991) High incidence of Tobacco streak virus in tobacco and its transmission by Microcephalothrips abdominalis and pollen from Ageratum houstonianum. Plant Dis 75:450–452
Shukla DD, Gough KH (1983) Tobacco streak, broad bean wilt, cucumber mosaic, and alfalfa mosaic viruses associated with ring spot of Ajuga reptans in Australia. Plant Dis 67:221–224
Sharman M, Thomas JE, Persley DM (2008) First report of Tobacco streak virus in sunflower (Helianthus annuus), cotton (Gossypium hirsutum), chickpea (Cicer arietinum) and mung bean (Vigna radiata) in Australia. Australas Plant Dis Notes 3:27–29
Sharman M, Persley DM, Thomas JE (2009) Distribution in Australia and seed transmission of Tobacco streak virus in Parthenium hysterophorus. Plant Dis 93(7):708–712
Almeida AMR, Sakai J, Hanada K, Oliveira TG, Belintani P, Kitajima EW, Souto ER, de Novaes TG, Nora PS (2005) Biological and molecular characterization of an isolate of Tobacco streak virus obtained from soybean in Brazil. Fitopatol Bras 30:366–373
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32(5):1792–1797. doi:10.1093/nar/gkh340
Maliogka VI, Dovas CI, Katis NI (2007) Demarcation of ilarviruses based on the phylogeny of RNA2-encoded RdRp and a generic ramped annealing RT-PCR. Arch Virol 152:1687–1698
Edwards JBDM, Delort J, Mallet J (1991) Oligodeoxyribonucleotide ligation to single-stranded cDNAs: a new tool for cloning 5’ ends of mRNAs and for constructing cDNA libraries by in vitro amplification. Nucleic Acids Res 19(19):5227–5232. doi:10.1093/nar/19.19.5227
Tessier DC, Brousseau R, Vernet T (1986) Ligation of single-stranded oligodeoxyribonucleotides by T4 RNA ligase. Anal Biochem 158(1):171–178
Langeveld SA, Dore J-M, Memelink J, Derks AFLM, van der Vlugt CIM, Asjes CJ, Bol JF (1991) Identification of potyviruses using the polymerase chain reaction with degenerate primers. J Gen Virol 72:1531–1541
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28(10):2731–2739. doi:10.1093/molbev/msr121
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688–2690. doi:10.1093/bioinformatics/btl446
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321. doi:10.1093/sysbio/syq010
Anisimova M, Gil M, Dufayard J-F, Dessimoz C, Gascuel O (2011) Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst Biol 60(5):685–699. doi:10.1093/sysbio/syr041
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26(19):2462–2463. doi:10.1093/bioinformatics/btq467
Candresse T, Morch MD, Dunez J (1990) Multiple alignment and hierachical clustering of conserved amino acid sequences in the replication-associated proteins of plant RNA viruses. Res Virol 141(3):315–329
Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Jackson JD, Ke Z, Lanczycki CJ, Lu F, Marchler GH, Mullokandov M, Omelchenko MV, Robertson CL, Song JS, Thanki N, Yamashita RA, Zhang D, Zhang N, Zheng C, Bryant SH (2011) CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res 39(suppl 1):D225–D229. doi:10.1093/nar/gkq1189
Bol JF (1999) Alfalfa mosaic virus and ilarviruses: involvement of coat protein in multiple steps of the replication cycle. J Gen Virol 80:1089–1102
Padidam M, Sawyer S, Fauquet CM (1999) Possible emergence of new geminiviruses by frequent recombination. Virology 265(2):218–225
Martin DP, Posada D, Crandall KA, Williamson C (2005) A modified Bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses 21:98–102
Smith JM (1992) Analyzing the mosaic structure of genes. J Mol Evol 34(2):126–129. doi:10.1007/bf00182389
Gibbs MJ, Armstrong JS, Gibbs AJ (2000) Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16(7):573–582. doi:10.1093/bioinformatics/16.7.573
Posada D, Crandall KA (2001) Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci 98(24):13757–13762. doi:10.1073/pnas.241370698
Cook G, de Miranda JR, Roossinck MJ, Pietersen G (1999) Tobacco streak ilarvirus detected on groundnut in South Africa. Afr Plant Prot 5(1):13–19
Tzanetakis IE, Mackey IC, Martin RR (2004) Strawberry necrotic shock virus is a distinct virus and not a strain of Tobacco streak virus. Arch Virol 149:2001–2011
Prasada Rao RDVJ, Reddy AS, Reddy SV, Thirumala-devi K, Chander Rao S, Manoj Kumar V, Subramaniam K, Yellamanda Reddy T, Nigam SN, Reddy DVR (2003) The host range of Tobacco streak virus in India and transmission by thrips. Ann Appl Biol 142:365–368
Acknowledgments
We thank Dr. David Teakle for providing a collection of archived isolates, including isolates -835, -837, -840 and -1025 used in this study. Dr. Ben Callaghan and Dr. Paul Campbell provided advice to enable sequencing of complete genome ends. Dr. Alistair McTaggart assisted with phylogenetic analysis. Mr. Denis Persley assisted with collection of field samples. This study was jointly funded by the Grains Research Development Corporation project DAQ00130 and the Cotton Research and Development Corporation project DAQ0002.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sharman, M., Thomas, J.E. Genetic diversity of subgroup 1 ilarviruses from eastern Australia. Arch Virol 158, 1637–1647 (2013). https://doi.org/10.1007/s00705-013-1628-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1628-4