
Abstract
In determining putative recombination events and their evolution rates in the RNAs 1 and 2 of currently the known members of the family Bromoviridae, a detailed study comprising 107 accessions retrieved from the international databases, has been carried out by using RECCO and RDP v3.31β algorithms. These programs allowed the detection of potential recombination sites in all the five virus genera composing the family Bromoviridae with various degrees of consistency. The RNAs 1 and 2 showed inferred phylogenies fully congruent and clearly delineated five clusters representing the five studied virus genera. In this respect, we proposed to classify the Ilarviruses in three distinct subgroups instead of 10 as mentioned in several reports of the International Committee on Taxonomy of Viruses where its suggestions were based on antigenic differences. Moreover, we confirmed that Alfalfa mosaic virus should be considered as a component of the Ilarvirus genus instead of being the unique representative of Alfamovirus genus. In addition, Pelargonium zonate spot and Olive latent 2 viruses fully deserve their affiliation to the family Bromoviridae.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Various factors have been involved in the emergence of new plant viruses including an expanded range of host and vectors, changes in climate and environment, new agricultural practices, and the general increasingly movement of humans populations and crops. One evolutionary process that might facilitate emergence by generating novel variants is recombination. Recombination, defined as the exchange of genetic information between two nucleotide sequences, is an important process that influences biological evolution at many different levels. Recombination explains a considerable amount of genetic diversity in natural populations and, in general, genes located in regions of the genome with low levels of recombination have low levels of polymorphism [31]. Recombination reshuffles existing variation and even creates new variants. A single virus isolate does not consist of a single RNA sequence, but of a population of related sequence variants, often referred to as quasispecies [9, 10, 16]. The quasispecies nature of RNA viruses implies a high adaptative potential, allowing for the rapid selection of biologically distinct sequence variants with the highest fitness in new environments. It may result in dramatic changes in the biological properties of the virus, with major epidemiological consequences, including the appearance of resistance-breaking strains or the acquisition of broader host ranges [22, 28]. It has been shown that RNA recombination enables the exchange of genetic material not only between the same or similar viruses but also between distinctly different viruses [41]. Sometimes, it also permits crossovers between viral and host RNA [1, 3, 15, 29]. Taking into account the structure of viral genomic molecules and the location of crossover sites, three basic types of RNA recombination were distinguished: Homologous, aberrant homologous, and non-homologous [2, 21]. The former two occur between two identical or similar RNAs (or between molecules displaying local homology), while the last one involves two different molecules. Most of the collected data suggest that RNA recombinants are formed according to a copy choice model [2]. A viral replication complex starts nascent RNA strand synthesis on one template, called RNA donor and then switches to another template, called RNA acceptor. Accordingly, two main factors are thought to affect RNA recombination: the structure of recombining molecules and the ability of the viral replicase to switch templates.
Amongst positive-strand plant RNA viruses, the family Bromoviridae encompasses several viruses having an important economical impact. According to the 8th ICTV report of the International Committee on Taxonomy of Viruses [11], the family Bromoviridae consists of five genera of plant viruses with a tripartite RNA: RNAs 1 and 2 encode the viral subunits of the replicase; RNA 3 is bicistronic and codes for a movement protein that is required for cell-to-cell movement (MP) and a coat protein (CP) needed for cell-to-cell and long-distance transport; CP is translated from a subgenomic messenger, RNA 4, that is coterminal with the 3′ 800–1,000 nucleotides (nt) of RNA 3. According to Tzanetakis and Martin [39], an additional open reading frame (ORF) was found in RNA 3 making Fragaria chiloensis latent virus (FCLV) probably the first Ilarvirus encoding a third protein via this genomic region.
There is a general belief that capsid protein genes of RNA viruses evolve much more rapidly than genes encoding components of the replication apparatus [18, 42]. Putative recombination events were detected in numerous CP genes of plant viruses namely in PNRSV [6]. Recently, we demonstrated that two additional viruses of perennial plants (Plum pox and Prune dwarf viruses) can exchange during recombination evolution of their CP genes, segments of genetic information exceeding 100 residues (unpublished data). Referring to RNAs 1 and 2 and even with some information provided by Codoner and Elena [8] regarding the occurrence of putative recombination events in these genomic regions, knowledge is still scarce especially in determining the recombination evolution rate along 107 examined sequences.
The aim of this study was threefold: (i) to detect potential recombination signals in all the RNAs 1 and 2 sequences of the different members of the family Bromoviridae available so far in the international databases; (ii) to provide a detailed study on their recombination evolution rate; (iii) to determine the evolutionary relationships among them.
Materials and methods
Virus sequence source
The sequences of RNAs 1 and 2 of 107 accessions used in this study were downloaded from GenBank (Table 1).
Sequence alignments, recombination, and phylogenetic analyses
The nucleotide sequences were aligned using CLUSTALW 2.0.9 and CLUSTALX 2.0.9 [23] with default parameters. Their phylogenetic relationships were determined by using the neighbor-joining (NJ) method implemented in MEGA4.1β program [20]. Bootstrap analyses with 500 replicates were performed to assess the robustness of the branches.
Potential recombination events between divergent nucleotide sequences were explored with two programs: RDP v3.31β [26] and RECCO [27]. RDP incorporates several published recombination detection methods into a single suite of tools: RDP [24], GENECONV [30], BOOTSCAN [25], MAXCHI [38], CHIMAERA [31], SISCAN [14], and 3SEQ [4]. In all these cases, defaults parameters were used. Only events predicted by half of the methods are considered as significant. The algorithm developed and described by Maydt and Lengauer, [27] a fast, simple and sensitive method for detecting recombination in a set of sequences and locating putative recombination breakpoints is based on cost minimization. This method has only two tunable parameters: recombination and mutation cost. In practice, the only parameter considered is α, representing the cost of mutation relative to recombination. When α changes from 0 to 1, the cost of mutation weighted by α increases, and the cost for recombination weighted by 1−α decreases. In other words, the parameter α controls the ambiguity between mutation and recombination.
Results
Recombination events during evolution of the Bromoviridae
Examination of the RECCO program output regarding the occurrence of recombination events in RNA 1 of the Bromoviridae, revealed that 13 out of 15 ilarviruses were putative recombinants (i.e., APLPV, CiLRV, EMoV, BCRSV, PMoV, TAMV, TSV, HJLV, FCLV, SPLV, ApMV, PDV, PNRSV). In contrast, CVV and SNSV did not show any recombinant signal (Table 2). RDP package gave the same results with, however, two exceptions: TAMV and TSV. Within the Ilarvirus genus, the most frequently recombining virus was APLPV (61 putative recombination sites), whereas the opposite was the virus EMoV (two sites). Alfalfa mosaic virus representing the single member of the genus alfamovirus is the most recombinant virus of the analyzed family Bromoviridae (75 sites), and its possible parental donors are EMoV and BCRSV, as the major and the minor parents, respectively. OLV-2 was recombinant also (55 sites), but RDP program did not provide any significant result. It was shown to be subjected to recombination with more than two-thirds of members of ilarvirus genus. Recombination investigations of the genus Bromovirus based on RECCO analysis, showed that only three members (BBMV, CYBV, SBLV) were recombinants (Table 3). These results were congruent with RDP package output except for CYBV. The only virus belonging to the proposed Anulavirus genus, i.e., PZSV was also recombinant with at least 34 potential recombination sites. Two isolates of PSV (Mild, and Rp) were recombining in 14 and 19 locations, respectively. Although RDP v3.31β algorithm did not confirm that PZSV and the Cucumovirus PSV (Mild, Rp) as recombinants, a strong support was given to four isolates of CMV (i.e., 42CM, CTL, P1-1, Tsh). In fact, most of the methods incorporated in RDP package confirmed the results obtained by RECCO and pointed out that TAV is the potential minor parental donor of no less than 17 CMV isolates. Seeking for the recombination evolution rate in RNA 1 of the Bromoviridae, the majority of Ilarvirus genus members showed that their breakpoint extent exceeded 3 nt but did not overstep 27 residues (APLPV) (Table 4). In contrast, the breakpoint extent of the greater number of putative recombination sites of AMV was limited to one single residue. The breakpoint extent in OLV-2 was nearly the same in 1, 2, 3,or >3 residues, but the largest size was the greater in the Bromoviridae RNA 1 (61 nucleotides). In majority, the breakpoint extent size of the Bromoviruses and Cucumoviruses overtook three nucleotides but not exceeding 54 residues (CMV.42CM) (Table 5).
Concerning RNA 2 of the Bromoviridae, all the studied ilarviruses were recombinants according to RECCO. RDP package confirmed these results except for APLPV, EMoV, BCRSV, PMoV, and PNRSV. Similarly, OLV-2 was revealed to be recombinant by RECCO but not by RDP 3.31β algorithm (Table 2). HJLV and APLPV were the highest recombinant viruses with 86 and 71 putative recombination breakpoints, respectively. OLV-2 and AMV belonging to two taxonomically distinct genera showed a high degree of recombination with 92 and 58 possible recombination sites, respectively. Recombination events occurred also in RNA 2 of the Bromoviruses (BBMV, CYBV, SBLV), PZSV, and all Cucumoviruses mentioned above supplemented, however, by seven more CMV isolates (i.e., pCb7, pRad35, China, BX, Palampur, pepo, AS) (Table 3). In determining potential recombination events, a high consistency was observed between RECCO and RDP package particularly for CMV and its various isolates as well as for TAV. Nonetheless, no confirmation was given by RDP program for the following viruses: BBMV, SBLV, PZSV, PSV.Mild, PSV.Rp. CMV.China, and CMV.BX. On the recombination evolution rate, approximately one half of members of Ilarvirus genus showed breakpoints extent consisting of one single residue particularly for HJLV (37 sites) and APLPV (29 sites) (Table 4). A similar situation was observed for OLV-2 (38 sites). Conversely, more than 80% of the recombinant Bromoviruses added to PZSV and Cucumoviruses displayed recombination sites having sizes overstepping three residues. The highest size was reached by CMV.P1-1 isolate (93 nt) (Table 5).
Nucleotide sequence analysis
Maximum composite likelihood estimate of the pattern of nucleotide substitution were conducted in MEGA4.1β. The results for the Bromoviridae were as follows: (i) RNA 1: rates of different transitional substitutions varied from 1.36 to 8.75, and those of transversionsal substitutions varied from 8.13 to 11.53. The nucleotide frequencies were as follows: 0.264 (A), 0.289 (T/U), 0.204 (C), 0.243 (G). The transition/transversion rate ratios were k 1 = 0.828 (purines) and k 2 = 0.168 (pyrimidines). The overall transition/transversion bias was R = 0.242, where R = [AGk 1 + TCk 2]/[(A + G)(T + C)]. There were a total of 2430 positions in the final dataset. (ii) RNA 2: rates of different transitional substitutions varied from 4.20 to 8.09, and those of transversionsal substitutions varied from 7.88 to 11.92. The nucleotide frequencies were as follows: 0.262 (A), 0.308 (T/U), 0.203 (C), 0.227 (G). The transition/transversion rate ratios were k 1 = 0.478 (purines) and k 2 = 0.679 (pyrimidines). The overall transition/transversion bias was R = 0.258. There were a total of 1750 positions in the final dataset.
The MEGA4.1β program implements also the Tajima’s Neutrality Test. The purpose of this test is to identify sequences which do not fit the neutral theory model at equilibrium between mutation and genetic drift. Tajima’s test compares a standardized measure of the total number of segregating sites (these are the DNA sites that are polymorphic) in the sampled DNA and the average number of mutations between pairs in the sample. The Tajima’s D was determined for RNA 1 (D = 4.567274) and RNA 2 (D = 4.677207).
Phylogenetic relationships
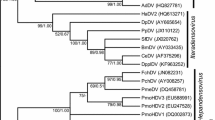
The phylogenetic relationships among members of the family Bromoviridae based on their RNAs 1 and 2 sequences were inferred using NJ method. As a general picture, RNAs 1 and 2 showed phylogenies fully congruent. Remarkably, each taxonomical genus in the family Bromoviridae constitutes a homogenous group clearly distinct from one another. Based on the inferred evolutionary history of members of Ilarvirus genus, AMV was shown to be an integral part of the genus Ilarvirus. In addition, we proposed to classify these members in three distinct subgroups (Fig. 1). Tentative subgroup I is formed by ApMV, PNRSV, FCLV, PDV, AMV, HJLV, and APLPV. Tentative subgroup II is composed of BCRSV, SNSV, TSV, and PMoV. Tentative subgroup III encompasses CiLRV, TAMV, SPLV, EMoV, and CVV. Clustering II and III are more closely related to each other among themselves than to any other member of the genus Ilarvirus.

Dendrogram depicting phylogenetic relationships among the studied members of the family Bromoviridae based on their RNA 1 and 2 sequences. Five clusters representing the five genera supplemented by the proposed Anulavirus genus, were clearly delineated. Three proposed subgroups forming the Ilarvirus genus were distinguished. The tree was produced using the N.J. algorithm option of MEGA4.1β. Bootstrap analysis of 500 replicates was performed. The scale bar shows the number of substitutions per nucleotide
Discussion
Studies of the molecular evolutionary history of viruses help to provide an understanding of important features of their biology such as changes in virulence and geographical ranges and their emergence as new epidemics, information that is essential for designing strategies for controlling viruses. Nonetheless, among evolutionary driving mechanisms, recombination can mislead the phylogenetic estimation procedure [32] and distort subsequent inferences based on inferred phylogenies [35, 36]. Consequently, an essential step in any phylogeny-based analysis is to screen for and quantify evidence for recombination [19]. In recent years, there has been an increased interest in understanding the role of recombination in the evolution of field populations of RNA plant viruses. Recombination may result in the exchange of long nucleotide sequences, and it could have bigger phenotypic effects than most mutations. This could jeopardize the efficiency of current control strategies, particularly so for the use of resistance to viruses bred in crop varieties [12]. In this study, we demonstrated the occurrence of putative recombination events in several members of the family Bromoviridae and determined their phylogenetic relationships. Several taxonomic implications can be drawn from the results obtained. First, as previously reported by various authors based on RNAs 1 and 2 sequences analysis [7, 33, 34, 37], AMV is basically separated from the ilarviruses primarily by its mode of transmission: it is transmitted non-circulatively by at least 14 species of aphids [17] and should not be considered as an independent genus but should be integrated in the genus Ilarvirus. Alfamovirus is not supported by molecular analyses and, thus, this genus does not correspond to a natural phenetic classification. Members of Ilarvirus genus should be classified into three distinct subgroups (Fig. 1) instead of 10 as mentioned in several reports of the International Committee on Taxonomy of Viruses where its suggestions were based on antigenic differences. As reported by Tzanetakis and Martin [39], serological relationships are not always reliable for assigning viruses into groups since some epitopes result from secondary and tertiary protein structures. For example, they mentioned that FCLV can be recognized by antisera of other Ilarviruses such as Lilac ring mottle and Asparagus 2 viruses. This clustering proposal agrees with the results provided by Codoner and Elena [7, 8]. Their results were based on the analysis of the whole genome as well as of the whole proteome of members of the family Bromoviridae. Second, PZSV which is the only current member of the proposed Anulavirus genus [13] deserves its affiliation to the Bromoviridae. Third, Oleavirus genus deserves too its affiliation to the family Bromoviridae. Nevertheless, our work showed a few inconsistencies with previous results provided by various workers. For instance, Shiel and Berger [34] stated that ApMV is more closely related to AMV than to other ilarviruses. This statement disagrees with our results which indicated that ApMV is more closely related to PNRSV than to AMV (Fig. 1). In fact, we determined here that PNRSV is the potential major parent of ApMV (Table 2). In addition and by contrast to Codoner and Elena [8], PNRSV was not a potential parental donor of AMV.
In biology, since the last few years until now, many applications have been based on the estimation of phylogenetic trees. One main assumption of numerous phylogenetic methods is that there is only one phylogeny underlying the evolution of the sequences under study. Recombination violates this assumption by generating mosaic genes, where different regions have different phylogenetic histories. By ignoring the presence of recombination, phylogenetic analysis may be severely compromised. Hence, the accurate detection of recombination from DNA sequences becomes very relevant, and indeed a number of methods have been developed for that purpose. Regarding RECCO, we showed in this article that it is capable to detect recombinants in all the studied genera. Likewise, it determined the rate of evolution of recombination events along the aligned sequences. In fact, using RECCO, we demonstrated in a previous study [6] that PNRSV can exchange during recombination evolution segments of the CP gene having a size as long as 100 residues contrary to, for instance, its RNAs 1 and 2 which exchanged only segments not exceeding 18 and four residues, respectively (Table 4). Similarly, PDV exchanged segments in the CP gene having a size of 196 nt, while its RNAs 1 and 2 did not exceed 21 and 17 residues, respectively (Table 4). Consequently, this is a clear indication that CP genes evolved more rapidly than genes coding for non-structural proteins. This is consistent with the statement of Zimmern [42] and Koonin and Gorbalenya [18]. By contrast, RDP package did not allow to reach such conclusions. In addition, the results obtained by RDP package were in some cases in line with those of RECCO, and in others not so. In our opinion, the main advantage of RDP 3.31β algorithm was to indicate the potential donators of the genetic information to recombinants. In a previous study [5], we dealt with PHYLPRO method [40] which demonstrated to be limited in that it was able to indicate only the position of the breakpoint represented by the value of the broken residue. Based on these observations, although RECCO is likely to give the most satisfactory results, both RECCO and RDP package should be considered as complementary methods, however.
Finally, to our knowledge, this article reports the largest study on recombination potentially occurring in the RNAs 1 and 2 of all the currently known members of the family Bromoviridae as well as their phylogenetic reconstruction.
References
R. Aaziz, M. Tepfer, J. Gen. Virol. 80, 1339–1346 (1999)
M. Alejska, A. Kurzyniska-Kokorniak, M. Broda, R. Kierzek, M. Figlerowicz, Acta Biochim. Pol. 48, 391–407 (2001)
M. Baroth, M. Orlich, H.J. Thiel, P. Becher, Virology 278, 456–466 (2000). doi:https://doi.org/10.1006/viro.2000.0644
M.F. Boni, D. Posada, M.W. Feldman, Genetics 176, 1035–1047 (2007). doi:https://doi.org/10.1534/genetics.106.068874
M. Boulila, Phytopathol. Mediterr. 46, 285–294 (2007)
M. Boulila, Plant Mol. Biol. Rep. (2008). doi: https://doi.org/10.1007/s11105-008-0071-2
F.M. Codoner, S.F. Elena, Arch. Virol. 151, 299–307 (2006). doi:https://doi.org/10.1007/s00705-005-0628-4
F.M. Codoner, S.F. Elena, J. Gen. Virol. 89, 1739–1747 (2008). doi:https://doi.org/10.1099/vir.0.2008/000166-0
E. Domingo, J. Holland, C. Biebricher, M. Eigen, in Molecular Basis of Virus Evolution, ed. by A.J. Gibbs, C.H. Calisher, F. Garcia-Arenal (Cambridge University Press, Cambridge, 1995), pp. 181–191
M. Eigen, Trends Microbiol. 4, 216–218 (1996). doi:https://doi.org/10.1016/0966-842X(96)20011-3
C.M. Fauquet, M.A. Mayo, J. Maniloff, U. Desselberger, L.A. Ball, Eighth Report of the International Committee on Taxonomy of Viruses (Elsevier/Academic Press, London, 2005)
F. Garcia-Arenal, B.A. McDonald, Phytopathology 93, 941–952 (2003). doi:https://doi.org/10.1094/PHYTO.2003.93.8.941
D. Gallitelli, M. Finetti-Sialer, G.P. Martelli, Arch. Virol. 150, 407–411 (2005). doi:https://doi.org/10.1007/s00705-004-0450-4
M.J. Gibbs, J.S. Armstrong, A.J. Gibbs, Bioinformatics 16, 573–582 (2000). doi:https://doi.org/10.1093/bioinformatics/16.7.573
A.E. Greene, R.F. Allison, Science 263, 1423–1425 (1994)
J.J. Holland, J.C. DeLaTorre, D.A. Steinhauer, in Genetic Diversity of RNA Viruses, ed. by J.J. Holland (Springer-Verlag, Berlin, 1992), pp. 1–20
E.M.J. Jaspars, L. Bos, in: CMI/AAB. Descriptions of Plant viruses, no 229 (1980)
E.V. Koonin, A.E. Gorbalenya, J. Mol. Evol. 28, 524–527 (1989). doi:https://doi.org/10.1007/BF02602932
S.L. Kosakovsky Pond, D. Posada, M.B. Gravenor, C.H. Woelk, S.D.W. Frost, Mol. Biol. Evol. 23(10), 1891–1901 (2006)
S. Kumar, M. Nei, J. Dudley, K. Tamura, Brief. Bioinform. 9(4), 299–306 (2008). doi:https://doi.org/10.1093/bib/bbn017
M.M.C. Lai, Microbiol. Rev. 56, 61–79 (1992)
J.P. Legg, J.M. Thresh, Virus Res. 71, 135–149 (2000). doi:https://doi.org/10.1016/S0168-1702(00)00194-5
M.A. Larkin, G. Blackshileds, N.P. Brown, R. Chenna, P.A. McGettigan, H. McWilliam, F. Valentin, I.M. Wallace, A. Wilm, R. Lopez, J.D. Thompson, T.J. Gibson, D.G. Higgins, Bioinformatics 23(21), 2947–2948 (2007). doi:https://doi.org/10.1093/bioinformatics/btm404
D. Martin, E. Rybicki, Bioinformatics 16, 562–563 (2000). doi:https://doi.org/10.1093/bioinformatics/16.6.562
D.P. Martin, D. Posada, K.A. Crandall, C. Williamson, AIDS Res. Hum. Retroviruses 21, 98–102 (2005). doi:https://doi.org/10.1089/aid.2005.21.98
D.P. Martin, C. Williamson, D. Posada, Bioinformatics 21, 260–262 (2005). doi:https://doi.org/10.1093/bioinformatics/bth490
J. Maydt, T. Lengauer, Bioinformatics 22(9), 1064–1071 (2006). doi:https://doi.org/10.1093/bioinformatics/btl057
F. Monci, S. Sanchez-Campos, J. Navas-Castillo, E. Moriones, Virology 303, 317–326 (2002). doi:https://doi.org/10.1006/viro.2002.1633
M. Nagai, Y. Sakoda, M. Mori, M. Hayashi, H. Kida, H. Akashi, J. Gen. Virol. 84(Pt 2), 447–452 (2003). doi:https://doi.org/10.1099/vir.0.18773-0
M. Padidam, S. Sawyer, C.M. Fauquet, Virology 265, 218–225 (1999). doi:https://doi.org/10.1006/viro.1999.0056
D. Posada, K. Crandall, Proc. Natl. Acad. Sci. USA 98, 13757–13762 (2001). doi:https://doi.org/10.1073/pnas.241370698
D. Posada, K.A. Crandall, J. Mol. Evol. 54, 396–402 (2002)
C. Rampitsch, K.C. Eastwell, Arch. Virol. 142, 1911–1918 (1997). doi:https://doi.org/10.1007/s007050050210
P.J. Schiel, P.H. Berger, J. Gen. Virol. 81, 273–278 (2000)
M. Schierup, J. Hein, Genetics 156, 879–891 (2000)
M. Schierup, J. Hein, Mol. Biol. Evol. 17, 1578–1579 (2000)
S.W. Scott, M.T. Zimmerman, X. Ge, Arch. Virol. 143, 1187–1198 (1998). doi:https://doi.org/10.1007/s007050050366
J.M. Smith, J. Mol. Evol. 34, 126–129 (1992)
I.E. Tzanetakis, R.R. Martin, Virus Res. 112, 32–37 (2005). doi:https://doi.org/10.1016/j.virusres.2005.02.010
G.F. Weiller, Mol. Biol. Evol. 15, 326–335 (1998)
M. Worobey, E.C. Holmes, J. Gen. Virol. 80, 2535–2543 (1999)
D. Zimmern, in RNA Genetics, ed. by J.J. Holland, E.R. Domingo, P. Ahlquist (CRC Press, Boca Raton, 1988), pp. 211–240
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Boulila, M. Recombination structure and genetic relatedness among members of the family Bromoviridae based on their RNAs 1 and 2 sequence analyses. Virus Genes 38, 435–444 (2009). https://doi.org/10.1007/s11262-009-0340-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-009-0340-7