
Abstract
Bovine leukemia virus (BLV)-infected cattle were classified by their proviral load into low and high proviral load profiles (LPL and HPL, respectively). Blood from these animals was used to infect sheep to obtain multiple identical copies of integrated provirus. An env fragment of BLV was amplified from all infected sheep and sequenced. The sequences that were obtained were compared to already published BLV genome sequence, resulting in three clusters. Mutations could not be attributed to the passage of provirus from cattle to sheep and subsequent amplification and sequencing. The description of two different proviral load profiles, the association of the BoLA-DRB3.2*0902 allele with the LPL profile, the availability of complete BLV sequences, and the comparison of a variable region of the env gene from carefully characterized cattle are still not enough to explain the presence of animals in every herd that are resistant to BLV dissemination.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bovine leukemia virus (BLV), family Retroviridae, genus Deltaretrovirus [1], is the causative agent of enzootic bovine leucosis (EBL) or lymphosarcoma. It is the most common neoplastic disease in cattle and causes major economic losses in cattle production and export [2, 3]. An NAHMS (National Animal Health Monitoring System, USA) study from 1996 to 2007 showed that herds with BLV produced $59 less in annual production per cow, or 3 % less milk, than non-BLV herds. Most of the BLV-infected cattle does not display clinical signs of the disease, and they are referred to as aleukemic or non-lymphocytotic (non-LP). Approximately 30 % of infected cattle develop a benign expansion of circulating B-cells, called persistent lymphocytosis (PL). Less than 5 % of infected cattle develop lymphosarcoma after an extended latency period of more than four years [4]. Bovines are the natural hosts of the virus. However, sheep are a convenient experimental model for studying pathogenesis of BLV infection because BLV-infected sheep show B-cell lymphoma and B-cell leukemia after a shorter latency period far more frequently than cattle [5]. Since BLV is a retrovirus, it integrates within host chromosomal DNA, where one to five copies of the provirus are usually found.
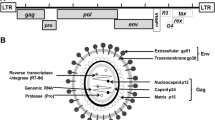
The provirus genome has the classical organization of a retrovirus. It is flanked by two identical long terminal repeats (LTRs) and contains the open reading frames (ORFs) corresponding to gag, pol and env, coding, respectively, for the viral capsid protein, the RNA-dependent DNA polymerase (the reverse transcriptase) and the envelope protein. Several ORFs, coding for Tax, Rex, R3 and G4 are present in the X region between env and the 3′ LTR. Tax and Rex are essential proteins required for transcriptional and post-transcriptional activation of viral expression. The R3 and G4 proteins are dispensable for infectivity but are involved in the maintenance of high viral loads [6].
A typical BLV infection is characterized by a high proviral load and strong and permanent antibody response against the two major antigenic viral proteins, gag p24 and the envelope protein gp51. p24 is a neutral and moderately hydrophobic protein, is the major constituent of the capsid of BLV virions, and appears to be the target for the host immune response, with high antibody titers found in the sera of infected animals. gp51 is a glycosylated protein that is located on the surface of viral particles and is the natural target for specific neutralizing antibodies. However, researchers have reported several cases of serologically negative cattle harboring the BLV provirus [7] and cases of serologically positive animals that do not harbor the BLV provirus. While in many cases, this could be an indication of the infection being in an early stage, in some other cases, it could indicate an atypical form of infection [8].
We have recently classified BLV-infected animals into two groups according to their proviral load and antibody titers against the structural proteins of BLV (gp51 and p24). The high proviral load profile (or HPL group) was characterized by the presence of more than 100,000 proviral copies integrated per microgram of peripheral blood leukocyte (PBL) DNA and high antibody titers against gp51 and p24. This constitutes a rather heterogeneous group, composed of lymphocytotic cattle and approximately 40 % of non-lymphocytotic cattle. The low proviral load profile (or LPL group) was characterized by an extremely low number of infected lymphocytes in peripheral blood (fewer than 100 copies of proviral DNA/μg of DNA) and normal peripheral blood lymphocytes counts. Although animals in this group had a rather strong immune response against gp51, they had a very low, sometimes undetectable response against p24. We and others have suggested that LPL cattle are not capable of transmitting BLV under normal dairy farm conditions, whereas HPL cattle (either those with PL or those with normal lymphocyte counts) are efficient BLV transmitters [9].
Currently, BLV is highly prevalent in several regions of the world [10]. After many years of culling infected animals, it has now been almost completely eradicated from the European Union [11, 12]. These onerous eradication programs are only possible in regions where viral prevalence is low or in countries where the government adopts economic reimbursement policies. Therefore, other strategies for eradication of the disease have also been considered, including isolation of infected animals, passive immunization with colostrum, and vaccination with viral proteins or attenuated strains, as well as some other less conventional approaches. However, none of these strategies currently achieve the optimal combination of efficiency, economy and safety [4].
Alternative approaches that target BLV control through genetic selection have been considered. We have recently found a strong association between certain alleles of the bovine leukocyte antigen (BoLA) DRB3.2* gene and the development of HPL or LPL. The alleles associated with the LPL profile, specifically *0902 and *1701, could be the markers of choice in the genetic selection of BLV-resistant cattle. The selection of BLV-resistant cattle by means of the aforementioned markers could represent an innovative tool for the control of BLV in heavily infected dairy farms, but the fact that the HPL profile was found in around 23 % of cattle carrying resistant alleles indicates that genetic resistance to BLV dissemination seems to occur by a more complex mechanism in which other genetic or epigenetic factors might be involved in the regulation of BLV infection, contributing to the outcome of the infection [13].
Some authors assume that small alterations in the env gene may affect infectivity and/or pathogenicity of BLV [14], considering that the BLV env glycoprotein plays a crucial role in viral infection and syncytium formation, since it is responsible for virus attachment and entry into host cells, serving as a target for neutralizing antibodies [15]. There is special interest in this gene, mostly because of this, but also because it is one of the most variable regions of the BLV genome [16] and it is subjected to immune responses and selection processes as a consequence of being a surface envelope protein. The in vivo mutation rate of the env gene, still considered very low, is approximately 0.009 % nucleotide changes per year [17]. It has been demonstrated that this gene contains several restriction sites that allow the classification of BLV strains into several different genotypes [15, 18]. Furthermore, it contains highly conserved regions that are involved in the interaction between target cells and the virus [19, 20]. Thus, minor changes in amino acid composition of the envelope protein could be responsible for differences in virus infectivity, BLV dissemination and progression of illness.
Our efforts are focused on finding other factors that could be implicated in the development of the high and low proviral load profiles, despite the genetic association mentioned above. The aim of this study was to analyze BLV isolates from cattle belonging to the two infection profile groups by comparing the nucleotide and amino acid sequences of a region of the env gene corresponding specifically to one neutralizing domain and the CD8+ T cell response epitope, as has been done by many other authors [7, 8, 15–22]. For this study, sheep were inoculated as a way to amplify the provirus isolated from the LPL group. As reported previously [23], this was an easily available alternative for the amplification of BLV provirus, mostly in the case of LPL cattle, in which the number of infected lymphocytes, and hence the number of proviral particles, is extremely low.
Materials and methods
Animal inoculation
Experimental animals were kept under natural rearing conditions on a private farm. Merino sheep and Argentinean Holstein cattle were used in this study. The management of experimental animals was done in accordance with institutional and internationally accepted welfare guidelines [24]. Inoculation of sheep was carried out by subcutaneous injection of whole blood from an infected donor. Comparable numbers of proviral copies were used for inoculation.
Sample collection
Ten ml of heparinized (5 U/ml) blood were obtained by jugular venipuncture. Plasma was harvested after centrifugation of blood samples for 20 min at 2,000 × g. Sodium azide was added (final concentration 0.2 %), and the plasma aliquots were stored at −20 °C until use. PBL were obtained by mixing the buffy coat with 11 ml of cold ammonium chloride buffer (150 mM NH4Cl, 8 mM Na2CO3, 6 mM EDTA) for one min to completely lyse the erythrocytes. The cell pellet, obtained by centrifugation at 1,000 × g for 7 min at 4 °C, was resuspended in 1 ml of phosphate-buffered saline (PBS), transferred to a 1.5-ml tube, and centrifuged at 10,000 × g for 2 min. The supernatant was discarded, and the PBLs were stored at −20 °C. Usually, more than 3 × 106 leukocytes were obtained from each sample. Animals were bled just before inoculation, and successive blood samples were taken from each animal every three months for a period of one year.
Determination of antibody titre against BLV gp51
The anti-BLVgp51 antibody titre was determined by testing twofold dilutions (from 1:50 to 1:6400) of plasma samples in a blocking immunoassay, designated ELISA108. The characteristics and evaluation of this ELISA have been reported [25].
Determination of antibody titre against BLV p24
The titre of antibodies against BLVp24 was determined in plasma samples by an ELISA, designated Rp24. The characteristics and evaluation of this assay, which employs a recombinant form of BLVp24 as antigen, have been previously reported [26].
PCR amplification
DNA was obtained from peripheral blood lymphocytes by a standard protocol [27]. To monitor BLV infection, a previously developed specific PCR with primers designed to detect BLV pol gene was perfomed [9].
DNA sequencing
A fragment of the env gene was amplified by nested PCR as described previously [8].
Forward and reverse primers were respectively env 5032 (5′-TCTGTGCCAAGTCTCCCAGATA-3′) and env 5099 (5′-CCCACAAGGGCGGCGCCGGTTT-3′), and env 5521 (5′GCGAGGCCGGGTCCAGAGCTGG-3′) and env 5608 (5′-AACAACAACCTCTGGGAGGGT-3′).
Amplification products were purified directly from the reaction tube using a PureLink™PCR Purification Kit (Invitrogen catalog no. K3100-01) according to the manufacter’s instructions. All of the amplified products were sequenced using the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit and an ABI377 Genetic Analyzer (Applied Biosystems).
Sequence analysis
In order to evaluate the genetic relationships between all of the isolates in this work and isolates for which a full genome sequence is available in the GenBank database, phylogenetic analysis was conducted. The analysis was carried out by the neighbor-joining method, using the MEGA (Molecular Evolutionary Genetics Analysis) software package version 4.0. The sequences were aligned using the ClustalW program.
Results
The proviral env region remains unchanged despite the change to a different host
Inoculation of sheep with BLV strains is a very common practice for amplifying BLV provirus from LPL cattle. However, it was necessary to determine that mutations did not occur in this host. To evaluate this issue, two sheep were inoculated with blood from a cow that belonged to the HPL group (#38) [28], and two other sheep were inoculated with blood extracted from an LPL animal (#41) [23]. Considering that animals in the LPL group carry approximately 100 copies of provirus per microgram of PBL DNA, while HPL animals carry at least 100,000 copies of provirus per microgram of PBL DNA [10], 100 ml of inoculum was needed to produce infection in the sheep host when inoculation was performed with blood from an LPL cow. When blood from an HPL cow was used for the inoculation, 1 microliter was enough to infect the animal. After three months, all sheep become positive for BLV infection, as determined by serology and PCR analysis. A region of the env gene from the sheep-integrated provirus was amplified by nested PCR and sequenced. No differences were found among the sequences obtained from the sheep and the donor cows (not shown). Thus, we assumed that the provirus did not mutate when cows were bled to inoculate sheep.
Env gene amplification and comparison of nucleotide and predicted amino acid sequences
BLV-infected cattle from six dairy herds from different regions of Argentina were used. Two animals, representing the HPL and LPL groups, were selected from each herd, and sheep were inoculated in duplicate. The experiment was designed to be independent from the genetic background of cattle, so specific care was taken to select cattle that did not harbour any of the alleles described to be associated with viral dissemination. Development of infection was monitored by serology and PCR assays. When animals seroconverted, a 400-nucleotide region of the BLV env gene was amplified by nested PCR and sequenced. Sequence alignments were performed using ClustalW (Fig. 1).

Multiple sequence alignment of partial sequences of gp51 from different isolates. Letters preceding the dash are a code for the specific dairy herd. HPL and LPL indicate high and low proviral load, respectively. “*” indicates nucleotide sequence identity. Numbers in the figure indicate the position of the referred sequence in the full-length genome (accession number AF257515), positions 5146-5545
Only minor sequence differences were found between the amplified regions of the proviral env gene derived from LPL and HPL cattle. These differences were randomly located in each group. Comparison at the amino acid level showed that four out of nine amino acid changes were detected in the “strong” conserved groups described by Dayhoff in 1978 [29], when developing the PAM250 matrix (Fig. 2). Theoretically, this finding indicates that these changes do not exert any effect on protein function.

Amino acid sequence alignment of a portion of the predicted gp51 protein. Comparison was performed using ClustalW. “*” (asterisk) indicates positions that have a single fully conserved residue. “:” (colon) indicates conservation between groups of strongly similar properties, scoring >0.5 in the Gonnet PAM250 matrix. “.” (period) indicates conservation between groups of weakly similar properties, scoring ≤0.5 in the Gonnet PAM 250 matrix. Epitopes and domains that were described previously are indicated by lines. The numbers in the figure indicate amino acid positions in the protein
Phylogenetic analysis of the obtained sequences
A phylogenetic tree was built by comparing the provirus sequenced in this work with the same region of published full-length BLV genomes (Fig. 3). Three clusters could be defined: one that comprises most of the sequences we have obtained, regardless of the group they belonged to, or the location they came from, another that includes only the Belgian strain, and a third that includes one sequence from the LPL group and one from the HPL group, both from different herds, and these also clustered with the Japanese and the Australian strains.

Phylogenetic tree based on partial nucleotide sequences of the BLV env gene from the different strains isolated for this work, compared to the same region of the other published full-genome sequences. The tree was generated by the neighbour-joining method, using 1000 bootstrap replications. Sa: Japanese, Au: Australian, Be: Belgian. Arg38 and Arg41 belong to the HPL and LPL group, respectively
Discussion
The most accepted techniques to detect BLV infection worldwide are AGID (agar gel immunodiffusion test) and ELISA against the 51-kD glycoprotein [30]. However, in the past decade, AGID has been almost completely replaced by the ELISA test. Both techniques have the drawback of giving false negative results, mainly because of the weak humoral immune response sometimes mounted to the infection, but also because of a lack of sensitivity of these techniques. Several studies have described the existence of a small group of animals that, despite being infected with BLV, do not exhibit the characteristic infection profile of a high proviral load and a strong immune response against the major structural viral proteins gp51 and p24. In some cases, these animals were considered infected by serology tests, but the integrated provirus was undetectable by PCR. In other cases, although antibodies were undetectable, the provirus could be amplified by PCR [7].
We have performed an extensive study in which a large number of infected animals (n=200) belonging to different dairy herds around the country were analyzed to detect BLV infection by direct PCR as well as by a blocking ELISA against gp51, and by another ELISA developed to detect antibodies against p24, both of which were developed in our laboratory [25, 26]. We found a relatively large number of animals that could be included in a distinctive group called low proviral load profile (LPL), with detectable antibody titers against gp51, very low or undetectable antibody titers against p24, and in most cases, negative results by direct PCR. BLV infection in those cases could be confirmed by a nested PCR or by real-time PCR (RT-PCR), but it is well known that the former is a very cumbersome technique that often yields false positive results, especially when one is working with a large number of samples, and the latter is expensive and not available in many laboratories.
An optional strategy to determine the sequence of the provirus of LPL animals is to amplify the viral strain in a more biologically susceptible host, such as sheep. However, when sheep are inoculated with a BLV provirus, the possibility exists that the provirus will undergo mutation while infecting lymphocytes of the new host. To demonstrate that mutations did not occur in our experiments, blood from BLV-infected cattle of the HPL category was inoculated into two sheep. The same procedure was done with blood from an LPL animal. A 400-bp region of the env gene was amplified and sequenced from the experimentally infected sheep, and the sequences that were obtained were compared to the corresponding sequence from the donor cows. No differences were found, indicating that at least the env region, which is involved in the interaction of the target cells and the virus [19, 20], remained constant.
Later, cattle belonging to the two distinguishable groups of high or low proviral load were selected from six different dairy farms located in different regions of Argentina. Sheep were inoculated with blood from one animal from each group. The region of the env gene described previously was amplified and sequenced from each infected sheep, considering the possibility that differences at the nucleotide level in that region could be associated with the development of the different infection profiles. No mutations that could be associated with any particular profile were detected, strengthening the hypothesis that the development of each profile could be associated primarily with some other genetic or epigenetic property of the host. It is interesting to note that at the protein level, nine amino acid differences were found among the sequences and that four of them were substitutions that theoretically do not imply a change in the biological activity of the protein according to the matrix analysis developed by Dayhoff [29].
Interestingly, comparative analysis indicated that the homology between all of the published full-length BLV genomes is greater than 96 % in all cases. In particular, a strain belonging to the high proviral load group (Arg38) and another from the low proviral load group (Arg41) were fully sequenced by our group [23, 28]. If we focus our attention on the LTR region, it contains the RNA transcription promoter and enhancer elements, the NF-KB binding site, the cyclic AMP response elements (CRE) and E box motifs, the PU box, the polyadenylation signal, the REX response element, and the proline tRNA primer-binding site typical of BLV. It plays an important role in viral transcription and hence in serological reactivity and viral load. There are very slight differences in this region when a comparison is made between Arg38 and Arg41, compared to other BLV genomes, which are not believed to be of functional significance. Only one amino acid change that occurs within the first enhancer region, at Arg38, could be responsible for the emergence of the two different infection profiles, although this is just an assumption; many tests have to be done to prove this hypothesis.
We have previously referred the presence of a single mutation, E161G, in the CD8+ T-cell epitope in the LPL strain [23] that, theoretically, could alter the stimulation of the anti-BLV CD8+ T-cell response [31]. This mutation did not appear in any of the env fragments analysed in this study.
These and other minor differences found among the available full BLV sequences are still not enough to fully explain the existence of two different infection profiles, so more studies related to the progression of viral infection in the host have to be done in which the host immune response is examined in more depth, together with the apoptotic/proliferation response elicited as a consequence of changes in the cytokine expression pattern [32].
When a phylogenetic tree was built based on a portion of the env region, derived from published full-length BLV genome sequences, including Arg38 (HPL profile) and Arg41 (LPL profile) as wells as the sequences obtained in this study, it was possible to group them into three clusters. Arg38 and Arg41 were located in one cluster, together with most of the selected strains, and this was independent of the localization of the herd or the infection profile to which they belonged. A second cluster was defined that included only the Belgian strain, and a third cluster, which comprised the Japanese and the Australian strains together with two of the selected animals, which had no geographical or familial relationship to each other.
A comparison of the amplified env regions of the animals analyzed in this study showed that the different isolates are randomly distributed in clusters. Strains did not cluster according to the infection profile they developed, while we could see some association due to the herd to which they belonged, suggesting that the same strain infects most of the animals in a herd, and this reinforces the idea that the development of one or another profile of infection is due to another genetic or epigenetic cause.
The description of two different profiles among infected cattle, the strong association of the allele BoLA-DRB3.2*0902 with the LPL profile, the publication of the full BLV sequences from cows with a high and low proviral load, and the comparison of a previously characterized region of the env gene from cattle are still not enough to fully explain the presence of animals in every herd that are resistant to BLV dissemination. Further studies are necessary in order to find a property of the hosts, the different strains of the BLV provirus, or a combination of several factors that completely explains the phenotype of these animals.
References
Fauquet CM, Mayo A, Maniloff J, Desselberger U, Ball LA (2005) Virus taxonomy, VIIIth report of the ICTV. Elsevier Academic Press, London
Ott SL, Johnson R, Wells SJ (2003) Association between bovine-leukosis virus seroprevalence and herd-level productivity on US dairy farms. Prev Vet Med 61:249–262
Rhodes JK, Pelzer KD, Johnson YJ (2003) Economic implications of bovine leukemia virus infection in mid-Atlantic dairy herds. J Am Vet Med Assoc 223:346–352
Rodríguez SM, Florins A, Gillet N et al (2011) Preventive and therapeutic strategies for bovine leukemia virus: lessons for HTLV. Viruses 3:1210–1248
Kabeya H, Fukuda A, Ohashi K, Sugimoto C, Onuma M (2001) Tumor necrosis factor alpha and its receptors in experimentally bovine leukemia virus-infected sheep. Vet Immunol Immunopathol 81(1–2):129–139
Florins A, Gillet N, Asquith B et al (2007) Cell dynamics and immune response to BLV infection: a unifying model. Front Biosci 1(12):1520–1531
Licursi M, Inoshima Y, Wu D et al (2002) Genetic heterogeneity among bovine leukemia virus genotypes and its relation to humoral responses in hosts. Virus Res 86:101–110
Fechner H, Blankenstein P, Looman AC et al (1997) Provirus variants of the bovine leukemia virus and their relation to the serological status of naturally infected cattle. Virology 237(2):261–269
Juliarena MA, Gutierrez SE, Ceriani C (2007) Determination of proviral load in bovine leukaemia virus-infected cattle with and without lymphocytosis. Am J Vet Res 68(11):1220–1225
Erskine R, Sordillo L (2009) Bovine leukosis virus update I: prevalence, economic losses, and management. Michigan Dairy Review, University of Michigan, MI
Nuotio L, Rusanen H, Sihvonen L, Neuvonen E (2003) Eradication of enzootic bovine leukosis from Finland. Prev Vet Med 59(1–2):43–49
Acaite J, Tamosiunas V, Lukauskas K, Milius J, Pieskus J (2007) The eradication experience of enzootic bovine leukosis from Lithuania. Prev Vet Med 82(1–2):83–89
Juliarena MA, Poli M, Sala L, Ceriani C et al (2008) Association of BLV-infection profiles with alleles of BoLA DRB3.2 gene. Anim Genet 39(4):432–438
Willems L, Kerkhofs P, Burny A, Mammerickx M, Kettmann R (1995) Lack of LTR and ENV genetic variation during bovine leukemia virus-induced leukemogenesis. Virology 206(1):769–772
Moratorio G, Obal G, Dubra A et al (2010) Phylogenetic analysis of bovine leukemia viruses isolated in South America reveals diversification in seven distinct genotypes. Arch Virol 155(4):481–489
Zaho X, Buehring G (2007) Natural genetic variations in bovine leukemia virus envelope gene: possible effects of selection and escape. Virology 360:150–165
Felmer R, Muñoz G, Zuñiga J, Recabal M (2005) Molecular analysis of a 444 bp fragment of the bovine leukaemia virus gp51 env gene reveals a high frequency of non-silent point mutations and suggests the presence of two subgroups of BLV in Chile. Vet Microbiol 108(1–2):39–47
Alkan F, Oğuzoğlu TC, Timurkan MO, Karapmar Z (2011) Characterisation of env and gag gene fragments of bovine leukemia viruses (BLVs) from cattle in Turkey. Arch Virol 156:1891–1896
Coulston J, Naif H, Brandon R et al (1990) Molecular cloning and sequencing of an Australian isolate of proviral bovine leukaemia virus DNA: comparison with other isolates. J Gen Virol 71(Pt 8):1737–1746
Mamoun RZ, Morisson M, Rebeyrotte N et al (1990) Sequence variability of bovine leukemia virus env gene and its relevance to the structure and antigenicity of the glycoproteins. J Virol 64(9):4180–4188
Camargos MF, Stancek D, Rocha MA, Lessa LM et al (2002) Partial sequencing of env gene of bovine leukemia virus from Brazilian samples and phylogenetic analysis. J Vet Med B49:325–331
Hemmatzadeh F (2007) Sequencing and phylogenetic analysis of gp51 gene of bovine leukemia virus in Iranian isolates. Vet Res Commun 31(6):783–789
Dube S, Abbott L, Dube DK et al (2009) The complete genomic sequence of an in vivo low replicating BLV strain. Virol J 6(1):120
American Veterinary Medical Association (2007) Guidelines for veterinarians and veterinary associations working with animal control and animal welfare organizations. Available from http://www.avma.org/issues/policy/comments/ofc_assc_guidelines_animal_control.asp
Gutiérrez SE, Dolcini GL, Arroyo GH, Rodriguez Dubra C, Ferrer JF, Esteban EN (2001) Development and evaluation of a highly sensitive and specific blocking enzyme-linked immunosorbent assay and polymerase chain reaction assay for diagnosis of bovine leukemia virus infection in cattle. Am J Vet Res 62(10):1571–1577
Juliarena M, Gutierrez S, Ceriani C (2007) Chicken antibodies: a useful tool for antigen capture ELISA to detect bovine leukemia virus without cross-reaction with other mammalian antibodies. Vet Res Commun 31(1):43–51
Sambrook J, Maniatis T (1989) Molecular cloning: a laboratory manual, vol 1. Cold Spring Harbor Laboratory Press, Woodbury, New York
Dube S, Dolcini G, Abbot L et al (2000) The complete genomic sequence of a BLV strain from a Holstein cow from Argentina. Virology 277(2):379–386
Dayhoff MO, Schwartz RM, Orcutt BC (1978) A model of evolutionary change in proteins. In: Dayhoff MO (ed) Atlas of protein sequence and structure, vol 5. Natl Biomed Res Found, Washington DC, pp 345–352
Choi KY, Liu RB, Buehring GC (2002) Relative sensitivity and specificity of agar gel immunodiffusion, enzyme immunosorbent assay, and immunoblotting for detection of anti-bovine leukemia virus antibodies in cattle. J Virol Methods 104(1):33–39
Gillet N, Florins A, Boxus M, Burteau C et al (2007) Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in humans. Retrovirology 4:18–49
Erskine RJ, Corl CM, Gandy JC, Sordillo LM (2011) Effect of infection with bovine leukosis virus on lymphocyte proliferation and apoptosis in dairy cattle. Am J Vet Res 72(8):1059–1154
Acknowledgments
We thank Dr. Sandra Perez for her helpful advice in the proofreading of this manuscript. The authors also thank Patricia Bani and Norma Rodriguez for technical assistance. This work has been partially supported by Consejo Nacional Investigaciones Cientificas y Tecnologicas (PIP 577) and Secretaria Ciencia, Arte y Tecnologia, Universidad Nacional del Centro de la Provincia de Buenos Aires.
Author information
Authors and Affiliations
Corresponding author
Additional information
M. A. Juliarena and P. A. Lendez contributed equally to the development of this work.
Rights and permissions
About this article
Cite this article
Juliarena, M.A., Lendez, P.A., Gutierrez, S.E. et al. Partial molecular characterization of different proviral strains of bovine leukemia virus. Arch Virol 158, 63–70 (2013). https://doi.org/10.1007/s00705-012-1459-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-012-1459-8