
Abstract
Autographa californica multiple nucleopolyhedrovirus (AcMNPV) odv-e25 is a core gene found in all lepidopteran baculoviruses, but its function is unknown. In this study, we generated an odv-e25-knockout AcMNPV and investigated the roles of ODV-E25 in the baculovirus life cycle. The odv-e25 knockout was subsequently rescued by reinserting the odv-e25 gene into the same virus genome. Fluorescence microscopy showed that transfection with the odv-e25-null bacmid vAcBacKO was insufficient for propagation in cell culture, whereas the ‘repair’ virus vAcBacRE was able to function in a manner similar to that of the control vAcBac. We found that odv-e25 was not essential for the release of budded viruses (BVs) into culture medium, although the absence of odv-e25 resulted in a 100-fold lower viral titer at 24 h post-transfection (p.t.). Analysis of viral DNA replication in the absence of odv-e25 showed that viral DNA replication was unaffected in the first 24 h p.t. Furthermore, electron microscopy revealed that polyhedra were found in the nucleus, while mature occlusion-derived viruses (ODVs) were not found in the nucleus or polyhedra in odv-e25 null transfected cells, which indicated that ODV-E25 was required for the formation of ODV.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Autographa californica multiple nucleopolyhedrosis virus (AcMNPV), a member of the genus Alphabaculovirus, family Baculoviridae, is the best-characterized baculovirus. Baculoviruses are arthropod-specific DNA viruses, and more than 600 viruses have been identified to date in a variety of insect orders [1, 13, 19, 22, 24, 33]. Baculovirus genomes are covalently closed circles of double-stranded DNA ranging from 80 to 180 k bp in size. Two viral forms, BV (budded virus) and ODV (occlusion-derived virus), are produced during the infection cycle. Even though the genomes of BV and ODV are genetically identical, the phenotypes of the two virions are structurally and functionally distinct [34]. The fact that ODV envelope proteins are more diverse in composition than those of BV envelope proteins determines the different roles of ODV and BV in viral infection [32]. BV is responsible for horizontal transmission of viral infection from cell to cell within a host, while ODV plays a critical role in vertical transmission of the virus from insect to insect.
Odv-e25 is conserved in all lepidopteran baculoviruses and is not found in other baculoviruses, such as dipteran NPVs and hymenopteran NPVs [34]. ODV-E25 is N-terminally anchored in the envelope, and the C-terminus interacts with the C-terminal region of ODV-E66 [5, 15, 30]. The AcMNPV odv-e25 (ORF94) is 687 bp long and predicted to code for a 228-aa (25.5 kDa) protein. A late transcriptional initiation motif (ATAAG) is found upstream of this gene. Some ODV envelope proteins contain a hydrophobic domain, such as ODV-E18, ODV-E56, and ODV-E66 [3, 4, 14], and the N-terminal 23 aa of ODV-E25 is conserved in all orthologs and directs the transport of ODV-E25 from the cytoplasm to the nucleus [15, 30].
Many ODV envelope proteins have been identified, such as odv-e26, odv-e66, p74, pif-1, pif-2, pif-3, and pif-4, and the functions of several of them have been investigated by deletion of the gene [7, 10, 18, 25, 26, 28]. ODV-E25, which has been identified as a structural protein of ODV that is also found on the BV envelope, is poorly understood because no gene deletion mutants have been generated [15, 29, 30, 38]. In this report, we generated an AcMNPV bacmid in which odv-e25 was disrupted by homologous recombination and investigated the role of odv-e25 in AcMNPV infection of Sf-9 cells using the odv-e25 null virus. The infectivity of the odv-e25-deletion virus was significantly reduced, and mature ODVs were not found in these virus-transfected cells.
Materials and methods
Cells
Sf-9 cells were maintained in TC-100 insect medium (Gibco) supplemented with 10% fetal bovine serum.
Construction of AcBacKO
Odv-e25 was deleted from an AcMNPV bacmid (AcBac), which was constructed according to the method reported by Xiang et al. [39] and maintained in DH10β using the λ Red recombination system (kindly provided by Dr Mary Berlyn, Yale University, USA) as described by Datsenko and Wanner [8]. In this bacmid, the polyhedrin gene is expressed under the p10 promoter. The chloramphenicol acetyl transferase (cat) gene (also provided by Dr. Mary Berlyn) with the arabinose promoter was amplified using primers 5′-tgaaattgtaaattaaaacaaatcatgtggggaatcgtgttactt gtgtaggctggagctgct-3′ and 5′-acaccaaagtgcaattagatttgcgcaattcgcgcacctctcgttcggcatatgaatatcctcctt-3′ (the nucleotides in the black box represent an artificially incorporated termination codon to terminate translation of the original product). Electrocompetent E. coli DH10β cells harbouring AcMNPV bacmid and pKD46 (λ Red recombinase genes) were transformed with 300 ng of the purified cat amplicon using an AxyPrep DNA Gel Extraction kit (Axygen). Cells were supplemented with 900 μl of fresh SOC medium and incubated for 3 h at 37°C with gentle shaking. Then, the cells were pelleted for 5 min at 5,000 rpm, resuspended in 150 μl of SOC broth, and plated on LB plates supplemented with kanamycin (50 μg/ml) and chloramphenicol (20 μg/ml). Colonies were subjected to PCR screening to confirm the deletion of odv-e25 and proper genomic insertion of cat. The colonies were identified by PCR (M13 F, 5′-tgtaaaacgacggccagt-3′ and M13 R, 5′-gaaacagctatgaccatgat-3′, and odv-e25 primers, odv-e25F, 5′-agtactatgtggggaatcgtg-3′ and odv-e25R, 5′-aggatccttaattcatttctctgta-3′).
gtgtaggctggagctgct-3′ and 5′-acaccaaagtgcaattagatttgcgcaattcgcgcacctctcgttcggcatatgaatatcctcctt-3′ (the nucleotides in the black box represent an artificially incorporated termination codon to terminate translation of the original product). Electrocompetent E. coli DH10β cells harbouring AcMNPV bacmid and pKD46 (λ Red recombinase genes) were transformed with 300 ng of the purified cat amplicon using an AxyPrep DNA Gel Extraction kit (Axygen). Cells were supplemented with 900 μl of fresh SOC medium and incubated for 3 h at 37°C with gentle shaking. Then, the cells were pelleted for 5 min at 5,000 rpm, resuspended in 150 μl of SOC broth, and plated on LB plates supplemented with kanamycin (50 μg/ml) and chloramphenicol (20 μg/ml). Colonies were subjected to PCR screening to confirm the deletion of odv-e25 and proper genomic insertion of cat. The colonies were identified by PCR (M13 F, 5′-tgtaaaacgacggccagt-3′ and M13 R, 5′-gaaacagctatgaccatgat-3′, and odv-e25 primers, odv-e25F, 5′-agtactatgtggggaatcgtg-3′ and odv-e25R, 5′-aggatccttaattcatttctctgta-3′).
Construction of vAcBac, vAcBacKO, and vAcBacRE
AcBacKO DNA and the helper plasmid pMON7124 were electroporated into E. coli DH10β cells, and subsequently, DH10AcBacKO competent cells were prepared. The enhanced green fluorescent protein gene (egfp) was amplified (primers: 5′-aatggatcctatggtgagcaag-3′ and 5′-cactagtgactagtcttgtac-3′) and cloned into pFastBacHTa, creating pFastBacHTa-egfp. Competent DH10AcBac cells (with helper plasmid pMON7124 and AcMNPV bacmid) and competent DH10AcBacKO cells were transformed with the recombinant plasmid, and the egfp gene was inserted into the polyhedrin locus by site-specific transposition. Thus, the constructed bacmids were subsequently identified and named vAcBac and vAcBacKO, respectively.
Similar to the SpltMNPV odv-e25 sequence [20], the AcMNPV odv-e25 sequence displayed a baculovirus consensus late transcriptional initiation motif (TTAAG) [2] at nucleotide residues −71 bp to −67 bp and one host factor binding site (GATA) [17] at nucleotide residues −77 bp to −74 bp with respect to the translational start codon. The whole odv-e25 gene (containing 327 bp upstream and 237 bp downstream of odv-e25) was amplified using primers (e25wh F: 5′-caagcttgtgaaagccttatttatg-3′; e25wh B: 5′-tttcgaacactcgtttgttcagtac-3′) and cloned into the pMD19-T vector (TaKaRa). The positive clone was identified and digested with Hind III, and e25wh was then cloned into the Hind III site of pFastBacHTa-egfp and pFastBscHTa, generating pFastBacHTa-egfp-e25wh and pFastBacHTa-e25wh. All of the recombinant plasmids used in this study were sequenced to confirm the correct form.
vAcBacRE was created by transforming competent DH10AcBacKO cells with pFastBacHTa-egfp-e25wh.
Transfection
Bacmid DNA was purified from 1.5 ml of LB cultures as described before [35] and detected using a NanoDrop ND-2000C. Cells (about 2 × 106/ml) were transfected with 2 μg of bacmid DNA using Cellfectin (Invitrogen) according to the manufacturer’s protocol. After 5 hours of incubation, the transfection mixture was removed, and the cells were gently washed twice with fresh TC-100 insect medium, and 2 ml of medium was added to cells.
Analysis of the viral growth curve
Virus supernatants were collected after cells were transfected with the bacmid at the given times (6, 12, 24, 48, 72, 96, and 120 h), and cell debris and cells were removed by centrifugation (5000 rpm for 5 min). An aliquot of each of these supernatants (250 μl) was processed using a viral DNA Kit (OMEGA). 2 μl of the viral DNA was used in triplicate to detect BV production by Q-PCR assay [21] with SYBR Premix Ex TaqTM (TaKaRa). A stock of control vAcBac (2.0 × 108 pfu/ml), previously titered by endpoint dilution and Q-PCR, was used as a standard and diluted tenfold. Q-PCR was performed by using a 7300 Real-Time PCR system (ABI) under the following conditions: 95°C for 30 s; 45 cycles of 95°C for 5 s and 60°C for 31 s, with a 500 μM concentration of each primer. A 50% tissue culture infective dose (TCID50) endpoint dilution assay was used to determine BV titers using Sf-9 cells in the 96-well microtiter plates.
Quantitative real-time PCR (Q-PCR) DNA replication assay
To detect viral DNA replication, a Q-PCR assay was performed as described previously [36, 37]. The primers gp41 F (5′-CGTAGTGGTAGTAATCGCCGC-3′) and gp41 R (5′-AGTCGAGTCGCGTCGCTTT-3′) were used to amplify a 101-bp region containing four Dpn I restriction sites within the gp41 ORF of AcMNPV. Sf-9 cells were transfected with vAcBac, vAcBacKO, and vAcBacRE and harvested at different time points. Total DNA was extracted using a Classic Genomic DNA Isolation Kit (Sangon) and digested with Dpn I (TaKaRa). Q-PCR was performed by using the 7300 Real-Time PCR system (ABI) under the following conditions: 95°C for 30 s; 45 cycles of 95°C for 5 s and 60°C for 31 s, with a 500 μM concentration of each primer.
Transmission electron microscopy
Sf-9 cells (2 × 106) were transfected with 6.0 μg of vAcBac, vAcBacKO, or vAcBacRE, and the cells were collected at 96 h post-transfection (p.t.) for transmission electron microscopy. The cells were first fixed with 2.5% glutaraldehyde in phosphate buffer (pH 7.0) for more than 12 hours, washed three times with phosphate buffer, and then once more for 15 min, postfixed with 1% OsO4 in phosphate buffer (pH 7.0) for 1 hour, and again washed three times with phosphate buffer. Then, the treated cells were first dehydrated using a graded series of ethanol (50%, 70%, 80%, 90%, 95%, and 100%) for about 15 to 20 minutes at each step, transferred to a mixture of alcohol and isoamyl acetate (V:V = 1:1) for about 30 minutes, and then transferred to pure isoamyl acetate for about one hour. Finally, the specimen was dehydrated in a Hitachi Model HCP-2 critical point dryer with liquid CO2. The dehydrated specimens were coated with gold-palladium and observed in a Philips Model XL30 ESEM.
Results
Construction and characterization of a recombinant bacmid
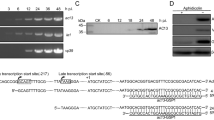
To determine the effect of odv-e25 knockout on virus replication, we generated an odv-e25-knockout AcMNPV bacmid via the λ Red homologous recombination system in E. coli. A sequence of 525 bp within the odv-e25 locus was replaced with a cat gene cassette (Fig. 1a). The precise deletion was confirmed by PCR; as expected, using primers odv-e25F/R, a PCR product of 1,212 bp was amplified from odv-e25-knockout bacmid vAcBacKO, a 696-bp fragment was amplified from vAcBac, and 696- and 1,212-bp fragments were confirmed from vAcBacRE (Fig. 1b). Using primers M13F/R, the PCR products of vAcBac, vAcBacKO, and vAcBacRE were 3,108, 3,108, and 4,359 bp, respectively (Fig. 1b). These results demonstrated that the recombinant bacmids used in this reports had been successfully constructed.

Schematic diagram and analysis of the bacmids used in this study. (a) Schematic diagram of the virus used in this study. Primers and replacement site are indicated by small black arrows. The open triangle and rectangle adjacent to cat are residues of odv-e25 that remained after recombination. (b) Identification of vAcBac, vAcBacKO, and vAcBacRE by PCR. The virus templates are indicated below each lane. In lanes 1, 3, and 5, the primer pair odv-e25F/R was used; in lanes 2, 4, and 6, the primer pair M13F/R was used. Marker sizes (bp) are indicated
Viral replication in Sf-9 cells
Sf-9 cells were transfected with vAcBac, vAcBacKO, or vAcBacRE and monitored by fluorescence microscopy. At 36 h p.t., no significant difference was observed among the cell lines transfected with vAcBac, vAcBacKO, or vAcBacRE, indicating comparable transfection efficiencies (Fig. 2a). The number of cells exhibiting fluorescence increased significantly after transfection with vAcBac and vAcBacRE, but in vAcBacKO-transfected cells, the number of infected cells scarcely changed. At 96 h p.t., the number of fluorescent cells transfected with vAcBac was equivalent to that of vAcBacRE, whereas there was no significant change in vAcBacKO -transfected cells (Fig. 2a). Supernatants from Sf-9 cells transfected with the three bacmids (120 h p.t.) were collected and used to infect 2 × 106 Sf-9 cells. At 36 h postinfection (p.i.), few cells infected with vAcBacKO were observed to emit fluorescence – far fewer than those infected with vAcBac/vAcBacRE. Fluorescence excitation was detected in nearly all cells infected by vAcBac/vAcBacRE at 96 h p.i., whereas there was no significant change in the number of vAcBacKO-infected cells (Fig. 2b).


Analysis of virus replication in Sf-9 cells. (a) Sf-9 cells were transfected with vAcBac, vAcBacKO or vAcBacRE and observed under a fluorescence microscope at 36 and 96 h p.t. (b) An infection assay was performed to detect replication and viral infectivity of vAcBac, vAcBacKO, and vAcBacRE in Sf-9 cells. (c) Brightfield images of vAcBac, vAcBacKO, and vAcBacRE infected cells at 96 h p.i. The arrows indicate cells containing polyhedra. (d) BV production curves of vAcBac, vAcBacKO, and vAcBacRE generated in a transfection time course experiment. (e) Examination of viral titre by TCID50 assay. (f) Quantitative analysis of replication of viral DNA. For (d), (e) and (f), error bars represent one standard deviation
(1) BV production and TCID 50 assays A virus growth-curve analysis was performed to determine the effect of odv-e25 on virus production. BVs were detected by Q-PCR to measure the extracellular viral DNA produced by cells transfected with each of the three bacmids. BV production was almost equivalent among the three bacmid-transfected cells from 6 to 24 h p.t. This suggested that the release of BVs from transfected cells was unaffected by the deletion of ODV-E25 (Fig. 2d). vAcBac- and vAcBacRE-transfected cells exhibited a steady increase in BV production, while that from cells transfected with vAcBacKO was almost constant starting at 24 h p.t. From 24 to 120 h p.t., the BV production of vAcBacKO increased about threefold, while that of vAcBac and vAcBacRE increased 1,000-fold. The production of BVs of vAcBac and vAcBacRE increased sharply from 24 to 72 h p.t. and reached a plateau at 72 h p.t.
A TCID50 endpoint dilution assay was used to detect infectious BVs. As expected, no infectious BVs were detected from cells transfected with the three bacmids transfected at 6 h p.t. The TCID50 of vAcBac and vAcBacRE was 2.37 × 104 and 1.78 × 104, respectively, at 24 h p.t., while that of the vAcBacKO was only 1.78 × 102. The viral titers of vAcBac and vAcBacRE were nearly 100 times higher than that of vAcBacKO (Fig. 2e). Considering that BV production was similar at 24 h p.t. with the three bacmids, the number of infectious viruses produced by the odv-e25 deletion virus was lower (Fig. 2f).
(2) DNA replication in Sf -9 cells To detect viral DNA replication in Sf-9 cells, total DNA was obtained from identical numbers of vAcBac-, vAcBacKO- and vAcBacRE-transfected Sf-9 cells and used for gp41-specific Q-PCR after digestion with Dpn I. From 6 to 24 h p.t., similar amounts of viral DNA were obtained from cells transfected with vAcBac, vAcBacKO and vAcBacRE, indicating that viral DNA synthesis was unaffected by the deletion of odv-e25 (Fig. 2f) during the first 24 h p.t. From 24 to 48 h p.t., the DNA accumulation of vAcBac and vAcBacRE increased 100-fold. In contrast, that of vAcBacKO was almost unchanged.
At 12 h p.i., BVs are produced by wt viruses, and until approximately 20 h p.i., production increases exponentially [16]. It is the infectious BVs that initiate secondary infection and lead to a continuous increase of DNA replication and BV production after 24 h p.i. Viral DNA replication has been detected before 12 h p.i. [31]. As reported before [30], the transcriptional and translational products of odv-e25 can be detected at 24 h p.i. (supplemental data). As expected, from 6 to 24 h p.t., the accumulation of vAcBac, vAcBacKO, and vAcBacRE DNA was similar. This indicated that all the three bacmids replicated in the transfected cells and that the deletion of ODV-E25 did not affect viral DNA replication during the first 24 h p.t. (Fig. 2f).
Transmission electron microscopic analysis of transfected cells
Electron microscopic analysis was performed to determine whether the deletion of odv-e25 had any effect on virus morphogenesis. Cells transfected with bacmid were harvested at 96 h p.t., and typical sections were chosen and analyzed. The results showed that typical ODVs were found in the nuclei and polyhedra of vAcBac- and vAcBacRE-transfected cells (Fig. 3a, c). However, there were no virions in the polyhedra or mature ODVs in the nuclei of vAcBacKO-transfected cells (Fig. 3b). For vAcBacKO, 15 cells were found to contain polyhedra in six typical sections, and 35 polyhedra were analyzed. In addition, nucleocapsids were found in all three bacmid-transfected cells, indicating that ODV-E25 was not required for the formation of the nucleocapsid (Fig. 3 and supplemental data).

Electron microscopic analysis of vAcBac (a, d, e), vAcBacKO (b, f, g), and vAcBacRE (c, h, i) bacmid-transfected Sf-9 cells at 96 h p.t. Panels a-c show micrographs of transfected cells. Panels d, f and h show higher-magnification micrographs of polyhedra. The presence of ODVs (white arrows) was found in vAcBac (e) and vAcBacRE (i) but not in vAcBacKO (g). The black triangles indicate nucleocapsids. P, polyhedron; C, cytoplasm; N, nucleus; NM, nuclear membrane; VS, virogenic stroma
Discussion
AcMNPV odv-e25 and its homologs are conserved in all lepidopteran NPVs and not found in dipteran and hymenopteran NPVs. The N-terminal portion of ODV-E25 is a transmembrane motif that is conserved in all homologs. Although found in BV, ODV-E25 is comparatively more abundant in ODV and is considered a structural protein of ODV [15]. However, the biological function of ODV-E25 remains mainly uncharacterized due to the inability to construct an odv-e25 knockout virus. In this study, we constructed an odv-e25 knockout bacmid using the λ Red recombination system. Our results show that ODV-E25 is required for the infectivity of BVs and the formation of ODVs.
The function of odv-e25 in the context of an AcMNPV infection in Sf-9 cells was analyzed with the odv-e25-null bacmid. The mutant virus could not be propagated after transfection (Fig. 2a, b), suggesting that BV production or viral infectivity was affected. To test this, BV production and TCID50 endpoint dilution assays were performed. The analysis showed that the deletion of odv-e25 affected infectious BV production. Comparison of the levels of viral DNA accumulation via Q-PCR in vAcBac-, vAcBacKO-, and vAcBacRE-transfected cells showed that viral DNA synthesis was unaffected by the deletion of odv-e25 in the first 24 h p.t. The difference between vAcBacKO and vAcBac/ vAcBacRE at 48 h p.t. is presumed to be caused by the different infectivity of BVs produced by the three bacmids. Polyhedra were found in odv-e25-null-infected cells (Figs. 2c, 3b), indicating that the expression of the polyhedrin gene and viral replication were unaffected [11]. On the other hand, it is concluded from our data that the defects caused by the deletion of odv-e25 were fully rescued by the ‘repair type’, and the replacement of odv-e25 by cat did not disturb the transcription of the DNA helicase gene, which is located downstream of odv-e25. The ODV-E25 protein appeared as a doublet with about 1 kDa separating the two bands (supplemental data), and a previous study showed that this protein does not appear to be glycosylated [30]. This suggested that another form of processing occurs, and further studies should be performed to see if these modifications affect the functions of ODV-E25.
Electron microscopy showed that nucleocapsids with a normal appearance were found in all three bacmid-transfected cells, indicating that nucleocapsid assembly was not affected by the deletion of odv-e25. The normal nucleocapsids also indicated that new viral DNAs were incorporated into capsids. This is consistent with the Q-PCR analysis, which showed that odv-e25 is not involved in viral DNA synthesis. Mature ODVs were found in the nuclei and polyhedra of vAcBac- and vAcBacRE-transfected cells (Fig. 3a, c) but were not found in vAcBacKO-transfected cells (Fig. 3b). This suggests that ODV-E25 is required for the formation of the mature ODV. Furthermore, ODV-E25 is considered to be part of the FP-25K/ODV-E66 complex [5, 7] and might play important roles in the functions of the complex and the procedure of ODV occlusion.
ODV envelope proteins play biological roles in ODV occlusion and interact with the midgut [32]. Current data indicate that up to 47 proteins are reported to be structural components of ODV [6, 9, 27]. Many of them are related to oral infection, such as PIF factors (PIF-1, PIF-2, PIF-3, and PIF-4) and do not affect BV infection [10, 12, 26, 28]. Some of ODV proteins have also been found in BVs, such as ODV-E18, which is essential for mediating BV production [23, 38]. AC109 is the first reported structural protein of the envelope and nucleocapsid from BV and ODV. In addition to being a structural protein of ODVs [30], ODV-E25 is suggested to be a structural component of BVs because it is required for producing infectious BVs. It has been demonstrated that the ODV-E25 N-terminal hydrophobic motif was sufficient to direct the protein through the nuclear membrane [15]. ODV-E25 is believed to be synthesized in the cytoplasm and transported into the nucleus to assemble the mature ODVs. However, it is not clear how ODV-E25 attaches to BVs. Additional studies should be performed to uncover the mechanism of action of ODV-E25 on BV infection and ODV formation.
References
Adams JR, McClintock JT (1991) Chapter6: Baculoviridae, nuclear polyhedrosis viruses Part 1. Nuclear polyhedrosis viruses of insects. 1:87–180
Blissard GW, Rohrmann GF (1990) Baculovirus diversity and molecular biology. Ann Rev Entomol 35:127–155
Braunagel SC, Elton DM, Ma H, Summers MD (1996) Identification and analysis of an Autographa californica nuclear polyhedrosis virus structural protein of the occlusion-derived virus envelope: ODV-E56. Virology 217:97–110
Braunagel SC, He H, Ramamurthy P, Summers MD (1996) Transcription, translation, and cellular localization of three Autographa californica nuclear polyhedrosis virus structural proteins: ODV-E18, ODV-E35, and ODV-EC27. Virology 222:100–114
Braunagel SC, Burks JK, Rosas-Acosta G, Harrison RL, Ma H, Summers MD (1999) Mutations within the Autographa californica nucleopolyhedrovirus FP25K gene decrease the accumulation of ODV-E66 and alter its intranuclear transport. J Virol 73:8559–8570
Braunagel SC, Russell WK, Rosas-Acosta G, Russell DH, Summers MD (2003) Determination of the protein composition of the occlusion-derived virus of Autographa californica nucleopolyhedrovirus. Proc Natl Acad Sci USA 100:9797–9802
Braunagel SC, Williamson ST, Saksena S, Zhong Z, Russell WK, Russell DH, Summers MD (2004) Trafficking of ODV-E66 is mediated via a sorting motif and other viral proteins: facilitated trafficking to the inner nuclear membrane. Proc Natl Acad Sci USA 101:8372–8377
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
Deng F, Wang R, Fang M, Jiang Y, Xu X, Wang H, Chen X, Arif BM, Guo L, Hu Z (2007) Proteomics analysis of Helicoverpa armigera single nucleocapsid nucleopolyhedrovirus identified two new occlusion-derived virus-associated proteins, HA44 and HA100. J Virol 81:9377–9385
Fang M, Nie Y, Harris S, Erlandson MA, Theilmann DA (2009) Autographa californica multiple nucleopolyhedrovirus core gene ac96 encodes a per Os infectivity factor (PIF-4). J Virol 83:12569–12578
Passarelli AL, Guarino LA (2007) Baculovirus late and very late gene regulation. Curr Drug Targets 8:1103–1115
Gutierrez S, Mutuel D, Grard N, Cerutti M, Lopez-Ferber M (2005) The deletion of the pif gene improves the biosafety of the baculovirus-based technologies. J Biotechnol 116:135–143
Herniou EA, Olszewski JA, Cory JS, O’Reilly DR (2003) The genome sequence and evolution of baculoviruses. Ann Rev Entomol 48:211–234
Hong T, Braunagel SC, Summers MD (1994) Transcription, translation, and cellular localization of PDV-E66: a structural protein of the PDV envelope of Autographa californica nuclear polyhedrosis virus. Virology 204:210–222
Hong T, Summers MD, Braunagel SC (1997) N-terminal sequences from Autographa californica nuclear polyhedrosis virus envelope proteins ODV-E66 and ODV-E25 are sufficient to direct reporter proteins to the nuclear envelope, intranuclear microvesicles and the envelope of occlusion derived virus. Proc Natl Acad Sci USA 94:4050–4055
Knudson DL, Harrap KA (1975) Replication of nuclear polyhedrosis virus in a continuous cell culture of Spodoptera frugiperda: microscopy study of the sequence of events of the virus infection. J Virol 17:254–268
Kogan PH, Blissard GW (1994) A baculovirus gp64 early promoter is activated by host transcription factor binding to CACGTG and GATA elements. J Virol 68:813–822
Kuzio J, Jaques R, Faulkner P (1989) Identification of p74, a gene essential for virulence of baculovirus occlusion bodies. Virology 173:759–763
Larsson R (1984) Insect pathological investigations on Swedish Thysanura: a nuclear polyhedrosis virus of the bristletail Dilta hibernica. J Invertebr Pathol 44:172–177
Li Z, Pan L, Yu H, Li S, Zhang G, Pang Y (2006) Identification and characterization of odv-e25 of Spodoptera litura multicapsid nucleopolyhedrovirus. Virus Genes 32:13–19
Lo HR, Chao YC (2004) Rapid titer determination of baculovirus by quantitative real-time polymerase chain reaction. Biotechnol Prog 20:354–360
Martignoni ME, Iwai PJ (1986) A catalog of viral diseases of insects, mites, and ticks. In: USDA Forest Service PNW-195, 4th edn. p 51
McCarthy CB, Theilmann DA (2008) AcMNPV ac143 (odv-e18) is essential for mediating budded virus production and is the 30th baculovirus core gene. Virology 375:277–291
Murphy FA, Fauquet CM, Bishop DHL, Ghabrial SA, Jarvis AW, Martelli GP, Mayo MP, Summers MD (1995) Classification and nomenclature of viruses: sixth report of the international committee for the taxonomy of viruses. Archives of Virology Supplement 10
O’Reilly DR, Passarelli AL, Goldman IF, Miller LK (1990) Characterization of the DA26 gene in a hypervariable region of the Autographa californica nuclear polyhedrosis virus genome. J Gen Virol 71(Pt 5):1029–1037
Ohkawa T, Washburn JO, Sitapara R, Sid E, Volkman LE (2005) Specific binding of Autographa californica M nucleopolyhedrovirus occlusion-derived virus to midgut cells of Heliothis virescens larvae is mediated by products of pif genes Ac119 and Ac022 but not by Ac115. J Virol 79:15258–15264
Perera O, Green TB, Stevens SM Jr, White S, Becnel JJ (2007) Proteins associated with Culex nigripalpus nucleopolyhedrovirus occluded virions. J Virol 81:4585–4590
Pijlman GP, Pruijssers AJ, Vlak JM (2003) Identification of pif-2, a third conserved baculovirus gene required for per os infection of insects. J Gen Virol 84:2041–2049
Rosas-Acosta G, Braunagel SC, Summers MD (2001) Effects of deletion and overexpression of the Autographa californica nuclear polyhedrosis virus FP25K gene on synthesis of two occlusion-derived virus envelope proteins and their transport into virus-induced intranuclear membranes. J Virol 75:10829–10842
Russell RL, Rohrmann GF (1993) A 25-kDa protein is associated with the envelopes of occluded baculovirus virions. Virology 195:532–540
Schultz KL, Friesen PD (2009) Baculovirus DNA replication-specific expression factors trigger apoptosis and shutoff of host protein synthesis during infection. J Virol 83:11123–11132
Slack J, Arif BM (2007) The baculoviruses occlusion-derived virus: virion structure and function. Adv Virus Res 69:99–165
Tinsley TW, Kelly DC (1985) Taxonomy and nomenclature of insect pathogenic viruses. In: Maramorosch K, Sherman KE (eds) Insecticides for biological control. Academic Press, USA, p 3
van Oers MM, Vlak JM (2007) Baculovirus genomics. Curr Drug Targets 8:1051–1068
Vanarsdall AL, Okano K, Rohrmann GF (2004) Characterization of a baculovirus with a deletion of vlf-1. Virology 326:191–201
Vanarsdall AL, Okano K, Rohrmann GF (2005) Characterization of the replication of a baculovirus mutant lacking the DNA polymerase gene. Virology 331:175–180
Vanarsdall AL, Okano K, Rohrmann GF (2006) Characterization of the role of very late expression factor 1 in baculovirus capsid structure and DNA processing. J Virol 80:1724–1733
Wang R, Deng F, Hou D, Zhao Y, Guo L, Wang H, Hu Z (2010) Proteomics of the Autographa californica nucleopolyhedrovirus budded virions. J Virol 84:7233–7242
Xiang X, Yang R, Yu S, Cao C, Guo A, Chen L, Wu X, Cui W, Cenis JL (2010) Construction of a BmNPV polyhedrin-plus Bac-to-Bac baculovirus expression system for application in silkworm, Bombyx mori. Appl Microbiol Biotechnol 87:289–295
Acknowledgments
This work was supported by National 973 Basic Research Program (2011) and Zhejiang Natural Science Foundation of China (Y3110058).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Chen, L., Hu, X., Xiang, X. et al. Autographa californica multiple nucleopolyhedrovirus odv-e25 (Ac94) is required for budded virus infectivity and occlusion-derived virus formation. Arch Virol 157, 617–625 (2012). https://doi.org/10.1007/s00705-011-1211-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-011-1211-9